
易错PCR提高粘质沙雷氏菌磷脂酶A1活性
2016-09-07 02:10王玉满薛正莲苏燕南安徽工程大学生物与化学工程学院安徽芜湖241000
安徽工程大学学报 2016年4期
王玉满,薛正莲,王 洲,苏燕南,朱 昊(安徽工程大学生物与化学工程学院,安徽芜湖 241000)
易错PCR提高粘质沙雷氏菌磷脂酶A1活性
王玉满,薛正莲∗,王洲,苏燕南,朱昊
(安徽工程大学生物与化学工程学院,安徽芜湖241000)
利用易错PCR技术对来自粘质沙雷氏菌PL-06的磷脂酶A1基因plaB进行定向改造,通过引入不同浓度Mg2+、Mn2+得到的易错PCR产物与表达型质粒p ET-28a(+)重组,构建磷脂酶A1突变文库,筛选出高酶活突变株EPB8.该突变酶相对野生酶酶活提高了22.5%,并对突变株EPB8产酶条件进行优化,优化后酶活较野生酶酶活提高41%.序列分析表明:plaB基因有11个碱基发生突变,导致7个氨基酸发生突变,其中磷脂酶A1蛋白有3个氨基酸突变,分别是Y97C、P98S、X228L,辅助蛋白有4个氨基酸突变,分别是P46L、Q58L、N136D、H233R.该实验结果为进一步在分子水平定向提高磷脂酶A1的活性奠定了基础,也为该酶在其他高表达系统中的表达提供了素材.
粘质沙雷氏菌;磷脂酶A1;易错PCR;产酶条件优化
磷脂酶A1是一类专一水解磷脂sn-1位酰基产生溶血磷脂和自由脂肪酸的酶类[1],可以用于植物脱胶和磷脂改性方面[2],其水解产物溶血磷脂是优良的乳化剂、食品添加剂和抗菌剂,广泛运用在食品、化妆品、医药等各个方面[3].随着磷脂酶的工业应用广泛,对其研究也日益增多.由于自然界筛选的菌株酶活不高、产量低,需进一步提高.随着分子技术水平的提高,与磷脂酶克隆表达相关的报道也随之增多.Michael Givskov[4]等提取液化沙雷氏菌磷脂酶A1基因在大肠杆菌克隆表达,但酶活不到1 U/m L.Jae Kwang Song[5]等构建基因文库成功克隆Serratia sp.MK中磷脂酶A1基因,并在大肠杆菌中表达,但表达量最高为2.21 ug/m L培养基.付建红[6]等将Serratia sp.xiF1内的磷脂酶A1基因分别在大肠杆菌和毕赤酵母中进行表达,在毕赤酵母中表达的重组酶在3.7 L发酵罐中磷脂酶A1活力达41.8 U/m L.但这些还不能满足工业应用的需求,因此,通过蛋白质工程手段对磷脂酶进行体外分子改造提高酶活性、稳定性及其产量具有重要意义.由于还未知磷脂酶的空间结构与功能的关系,故研究采用易错PCR技术改造磷脂酶的分子结构.易错PCR可以体外筛选突变蛋白达到迅速提高酶活力的目的,同时改善酶的一系列性质[7].王颖秋[8]等运用易错PCR方法提高了头孢菌素C酰化酶活力.刘韩[9]等运用易错PCR方法提高土芽孢杆菌ZH1羧酸酯酶的热稳定性.Stephens[10]等运用易错PCR方法改善了真菌木聚糖酶的耐碱性和耐热性.研究以粘质沙雷氏菌PL-06基因组为模板,扩增得到含辅助蛋白的磷脂酶A1基因plaB为研究对象,以期通过易错PCR法提高酶活.
1 材料与方法
1.1材料
(1)菌株和质粒.粘质沙雷氏菌PL-06由安徽工程大学微生物实验室筛选保存;宿主菌大肠杆菌BL21(DE3)、JM109、质粒pET-28a由安徽工程大学微生物实验室保存;工程菌BP28/BL21(plaB-p ET-28a/ BL21(DE3))由安徽工程大学微生物实验室构建保存.
(2)酶和试剂.PCR系列试剂、质粒DNA小量抽提试剂盒、DNA胶回收试剂盒、PCR产物纯化试剂盒、酶等购自上海生工生物工程技术服务有限公司;GeneRuler DNA Ladder Mix、T4 Ligase、限制性内切酶购自Thermo,其他试剂均为国产分析纯;引物合成由上海英潍捷基贸易有限公司合成.
(3)培养基.LB培养基:酵母粉5 g(0.5%)、胰蛋白胨10 g(1%)、NaCl 10 g(1%)加水至1 L,p H调至7.0,固体培养基加入2%琼脂粉;PLB培养基:卵磷脂20 g(2%)、胰蛋白胨10 g(1%)、NaCl 10 g(1%)、酵母粉5 g(0.5%)、0.02%溴甲酚紫(3%)、25 mmol/L CaCl2、1 000 m L蒸馏水,p H调至7.0;卵磷脂用高速匀浆机乳化后单独灭菌,固体培养基加入2%琼脂粉;卡那霉素(Kna)储存质量浓度为50 mg/m L,其工作质量浓度为50 ug/m L.
1.2方法
(1)易错PCR法扩增磷脂酶A1(plaB)基因.以粘质沙雷氏菌PL-06基因组为模板,利用易错PCR对目的基因引入突变,目的基因的上游引物:(划线部位为Bam HⅠ的酶切位点),下游引物为(划线部分为HindⅢ的酶切位点).50 u L的易错PCR反应体系:5 u L 10×PCR Buffer(无Mg2+)、7 mmol/L MgCl2、0.1 mmol/L MnCl2、5 U Taq DNA Polymerase、上下游引物各10 pmol、模板DNA 20 ng,补dd H2O至50 u L.反应程序为95℃5 min,95℃30 s,55℃30 s,72℃2 min,72℃8 min,35个循环,PCR产物检测后并回收,进行下一步实验.
(2)突变文库的构建.将纯化后易错PCR产物和载体pET-28a质粒用限制性内切酶Bam HⅠ、HindⅢ进行消化,经过T4连接酶连接后,随即转入宿主菌JM109感受态细胞中,涂布于带有卡那霉素(50 ug/m L)的LB平板上,37℃倒置培养12~16 h.将平板上长出的菌落接种至含有卡那霉素(50 ug/m L)培养液的LB培养基中,37℃培养12 h后提取质粒,利用化学转化法转入宿主菌BL21(DE3)感受态细胞,涂布于含有卡那霉素(50 ug/m L)的PLB平板上,37℃倒置培养12~16 h,构建突变文库.
(3)磷脂酶A1酶活突变株的初筛.PLB平板含有卵磷脂、溴甲酚紫和CaCl2,可作为筛选平板,产磷脂酶克隆会出现水解晕圈,将含有磷脂酶A1基因plaB重组质粒的工程菌BL21(DE3)点种在筛选平板上作为对照,根据圈径比的大小,初步筛选出正向突变株.
(4)目的菌株的复筛及鉴定.将初筛得到的单菌落接种至5 m L含有卡那霉素(50 ug/m L)培养液的LB培养基中,37℃、200 r/min培养12 h,培养物按2%的接种量接至100 m L含有卡那霉素的LB培养液中,37℃、200 r/min培养至菌浓为OD600=0.6(对数生长期)时,加入终浓度为0.2 mmol/L的IPTG,37℃诱导4 h,取发酵液,采用酸碱滴定法[11].酶活力定义为:在45℃和p H 6.0条件下,1 g固体酶粉(或1 m L液体酶)1 min水解磷脂产生1μmol的可滴定脂肪酸,即为1个酶活力单位(U/g或U/m L)[12],测定发酵液酶活.选取酶活提高的菌株,提取其质粒经限制性酶酶切验证后进行测序分析.
(5)突变菌株EPB8诱导生长曲线的测定.从平板上挑取P28(p ET-28a(+)/BL21(DE3))、BP28(BP28/BL21(DE3))、EPB8单菌落,接种于5 m L含卡那霉素(50 ug/m L)的LB培养液中,37℃、200 r/min过夜培养.1%的接种量,转接至100 m L含卡那霉素(50 ug/m L)的LB培养液的250 m L三角瓶中,37℃、200 r/min培养3~4 h,OD600为0.6时加入终浓度为0.2 mmol/L IPTG开始计时,每隔1 h取样测OD600值,记录数据绘制生长曲线.
(6)EPB8诱导产酶条件的优化.接种1 m L过夜活化的EPB8菌液至100 m L LB培养基中,分别从诱导时间、诱导温度、加入IPTG时的OD600值、IPTG浓度4个方面对EPB8进行产酶条件优化[12].诱导时间分别为2 h、4 h、6 h、8 h、10 h、12 h;诱导温度分别为22℃、27℃、32℃、37℃、42℃;加入IPTG时的OD600值分别为0.1、0.3、0.5、0.8、1.0、1.2;IPTG加量分别为0.05 mmol/L、0.1 mmol/L、0.2 mmol/L、0.3 mmol/L、0.5 mmol/L、0.7 mmol/L、1.0 mmol/L;诱导后进行酶活定量测定.初始时IPTG加量0.2 mmol/L、温度37℃、OD600值0.6条件下进行优化即可.
2 结果与分析
2.1目的基因的获得
将粘质沙雷氏菌PL-06接种至5 m L的LB培养液中,37℃、200 r/min培养12~16 h,所得菌液按照基因组提取试剂盒说明步骤进行操作,得到的基因组用1%琼脂糖凝胶验证进行.
2.2易错PCR条件的确定
由于Mg2+可以稳定非互补碱基对,因此增加Mg2+浓度使之超过正常用量,从而稳定非互补的碱基对.而Mn2+能够降低聚合酶对模板的特异性,提高错配率[13].研究通过改变Mg2+浓度和添加Mn2+促进碱基的错配,获得多样性突变文库.采用Mg2+浓度为1 mmol/L、3 mmol/L、5 mmol/L、7 mmol/L,Mn2+浓度为0、0.1 mmol/L、0.2 mmol/L、0.3 mmol/L、0.4 mmol/L、1 mmol/L作为梯度进行易错PCR.
不同离子浓度下的易错PCR如图1所示.由图1可知,随着Mn2+浓度的增加,目的基因扩增量减少,条带弥散性也较严重,所以最适Mn2+浓度为0.1 mmol/L.在最适Mn2+浓度下,Mg2+浓度过低,扩增效率低,甚至没有条带,若Mg2+浓度较低,突变率会较低[14],因而选择较高的Mg2+浓度7 mmol/L.不仅如此,条件6组合浓度基因平均突变率为0.58%,条件8组合浓度为0.63%,所以最终选择Mg2+浓度为7 mmol/L、Mn2+浓度为0.1 mmol/L,从而实现对目的基因突变率的有效地控制.
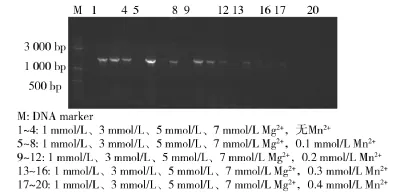
图1 不同离子浓度下的易错PCR
2.3基因文库的构建
按照上述易错PCR的条件,以基因组为模板扩增目的基因,大小约1 700 bp,易错PCR扩增目的基因如图2所示.经胶回收后,同时和载体pET-28a进行双酶切后连接,转化宿主菌大肠杆菌JM109感受态细胞中得到的转化子构成突变文库.
2.4突变株的筛选
(1)初筛.将文库中的转化子接种至LB培养液中混合过夜培养,提取质粒并转化至宿主菌大肠杆菌BL21(DE)感受态细胞中,涂布在含卡那霉素的PLB平板上,37℃培养12~14 h后,利用磷脂酶A1可以分解卵磷脂在PLB平板产生水解晕圈,根据水解晕圈的大小及其产生的快慢,可以直观地筛选酶活突变株.将筛选出酶活发生变化的突变株点种在含卡那霉素的PLB平板上,与BP28/BL21(简称BP28)比较晕圈大小,结果如图3所示.由图3可知,挑取圈径比较大的突变子6#、8#、16#、18#、19#、21#,在此基础上进一步复筛.
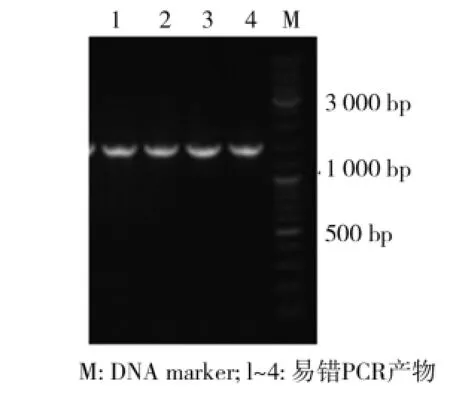
图2 易错PCR扩增目的基因

图3 PLB平板筛选结果
(2)复筛.将初筛得到的酶活改变的突变株接种至含有50 ug/m L卡那霉素的LB培养中,37℃、200 r/min进行培养至OD600为0.6时,加IPTG诱导4 h,测发酵液粗酶液酶活进行复筛,结果如图4所示.由图4可知,酶活都有所提高,但8#突变子提高幅度最大,酶活为19.6 U/m L.为进一步验证突变子的正确性,对其提取质粒进行单酶切验证,结果如图5所示.从图5中可以看出,突变株单酶切的大小约为7 000 bp和质粒(约5 300 bp)与目的基因(约1 700 bp)共同片段的大小相一致.条带大小正确说明突变子构建成功,并命名为EPB8以备下一步实验.
2.5突变基因的序列分析
提取突变株的重组质粒经PCR及酶切验证正确后,送上海生工生物有限公司进行测序.对比测序结果并分析突变率,找出碱基突变和相应氨基酸突变位点.
突变基因的序列分析表明,突变菌株EPB8有11个碱基发生突变,导致有7个氨基酸发生突变.其中,磷脂酶A1蛋白有3个氨基酸突变,分别是Y97C、P98S、X228L;辅助蛋白有4个氨基酸突变,分别是P46L、Q58L、N136D、H233R.
2.6突变株EPB8诱导生长曲线的测定
按1.2(5)所述方法进行诱导生长曲线的测定,结果如图6所示.由图6可知,BP28和突变株EPB8在未诱导的情况下,生长情况基本相一致.但加入IPTG诱导后,两菌株生长均表现出受到抑制.一方面IPTG对菌体有一定毒害作用;另一方面,目的产物磷脂酶A1对细胞也有一定的毒性[12].

图4 突变株摇瓶酶活测定
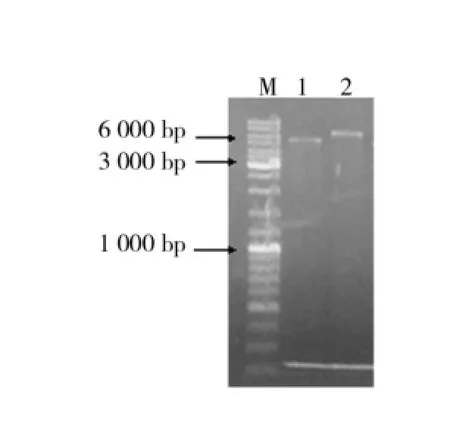
图5 突变菌株的酶切图谱

图6 EPB8突变株的诱导生长曲线
2.7突变菌株EPB8产酶条件的优化
(1)诱导时间对产酶的影响.按1.2(6)所述方法进行测定,结果如图7所示.由图7可知,诱导时间2 h时酶活性最高,随着时间的延长,酶活性有所降低,可能IPTG和磷脂酶A1对菌体都有一定毒害作用.诱导时间不宜过长,过长则菌体可能发生自溶,从而影响目的产物表达.故EPB8菌株最佳诱导时间为2 h.
(2)诱导温度对产酶的影响.诱导温度对目的产物的表达及酶活有着至关重要的作用,若诱导温度过高,重组蛋白会形成大量的无活性包涵体,以致胞外酶活降低;温度过低,影响菌体生长,从而影响重组蛋白的表达.按照1.2(6)所述方法进行测定,诱导2 h测定发酵液酶活,结果如图8所示.由图8可知,诱导温度为37℃时,酶活性最高,诱导温度过高或过低,酶活性都会降低,所以,最佳诱导温度为37℃.
(3)诱导起始OD600对产酶的影响.诱导起始OD600对菌体产酶有着较大影响,按照1.2(6)所述方法测定,诱导温度为37℃、诱导时间2 h后测定发酵液酶活,结果如图9所示.由图9可知,诱导时间过早,菌体生长缓慢且菌体量少,外源蛋白的表达量可能会降低,酶活性也不高;若诱导时间过迟,细菌自身代谢能力降低,不利于外源基因表达,酶活性也是相对较低.当OD600为0.8时诱导,磷脂酶A1活性最高,诱导起始OD600过高过低都会影响酶活,所以最佳诱导起始OD600为0.8.
(4)IPTG诱导浓度对产酶的影响.按照1.2(6)所述方法确定IPTG最佳诱导浓度,诱导温度为37℃、诱导起始OD600为0.8、诱导2 h后测定发酵液粗酶活,结果如图10所示.由图10可知,IPTG浓度为0.3 mmol/L时,酶活性最高,IPTG作为诱导剂能启动lac启动子的转录,使目的基因得以表达[12];IPTG浓度>0.3 mmol/L时,酶活性降低,可能是IPTG本身对细胞具有一定的毒性,对菌体生长有一定抑制作用,导致蛋白质产量降低;IPTG浓度<0.3 mmol/L时,酶活性也降低.因此,确定最佳的IPTG诱导浓度为0.3 mmol/L.

图7 诱导时间对酶活的影响

图8 温度对酶活的影响

图9 诱导OD600对酶活的影响
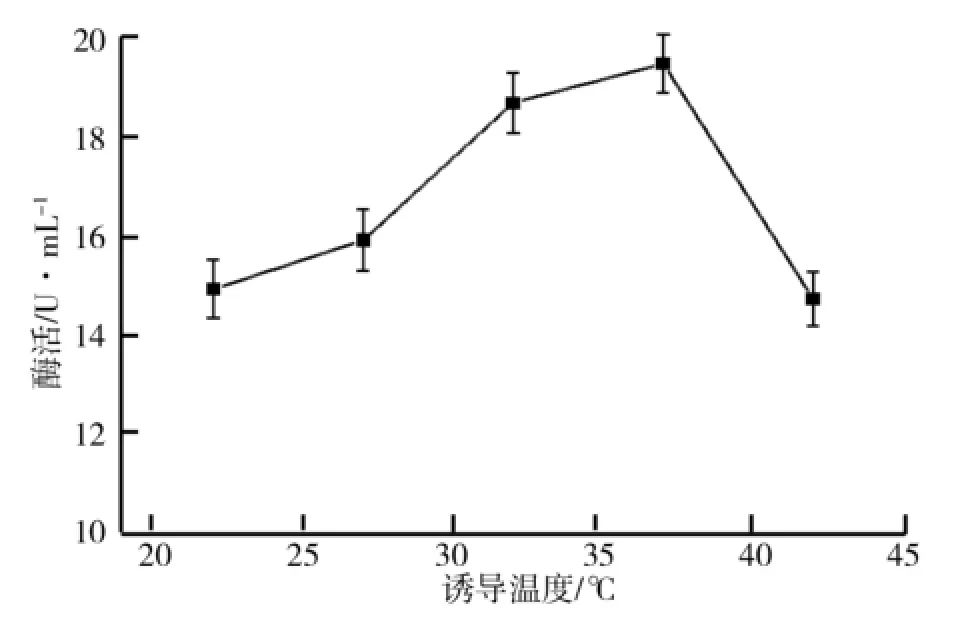
图10 IPTG浓度对酶活的影响
3 结论与讨论
研究确定了易错PCR条件:Mg2+浓度为7 mmol/L、Mn2+浓度为0.1 mmol/L,成功构建了突变文库.通过筛选最后得到酶活提高的突变株EPB8,该突变株最初酶活性测定为19.6 U/m L,相对于未突变前菌株BP28的酶活16 U/m L提高了22.5%,通过进行单因素实验优化,优化后的酶活达22.5 U/m L,比优化前提高了14.8%.
粘质沙雷氏菌磷脂酶A1基因plaB经易错PCR方法的定向改造后,磷脂酶A1活性有了较大提高,为后期磷脂酶结构与功能的关系研究提供了一定的素材.突变株EPB8经测序分析表明:有7个氨基酸发生突变,其中288位由X突变为L,与之前推断的第195位Ser为此磷脂酶A1的催化中心相距较近,这也可能是酶活提高的原因之一;而亮氨酸又为疏水性氨基酸,疏水性氨基酸在蛋白质内部,具有保持蛋白质的三级结构的作用,288位氨基酸的突变可能会使磷脂酶A1的三级结构更加稳定,这也为磷脂酶A1的研究奠定了一定的基础.
[1]G S Richmond,T K Smith.Phospholipases A1[J].Molecular Sciences,2011,12(1):588-612.
[2]杨博,王洪建.经济环保的酶法脱胶技术[J].中国油脂,2004,29(3):21-23.
[3]L De Maria,J Vind,K M Oxenboll,et al.Phospholipases and Their Industrial Applications[J].Applied Microbiology and Biotechnology,2007,74(1):290-300.
[4]G Michael,O Lars,M Soren.Cloning and Expression in Escherichia Coli of the Gene for Extracellular Phospholipase A1 from Serratia Liquefaciens[J].Journal of Bacteriology,1988,170(12):5 855-5 862.
[5]J K Song,J S Rhee.Simultaneous Enhancement of The rmostability and Catalytic Activity of Phospholipase A1 by Evolutionary Molecular Engineering[J].Applied and Environmental Microbiology,2000,66(3):890-894.
[6]J H Fu,H Q Huang,K Meng.A Novel Cold-Adapted PLA1 from Serratia sp.xjF1:Gene Cloning,Expression and Characterization[J].Enzyme and Microbial Technology,2008,42(2):187-194.
[7]张赛,邢克克,胡亚冬,等.易错PCR的黄曲霉毒素解毒酶体外分子定向进化[J].生物工程学报,2011,27(7):1 100-1 108.
[8]王颖秋,郑林冲,谢丽萍,等.基于易错PCR的头孢菌素C酰化酶的定向进化[J].中国医药工业杂志,2013,44(4):344-347.
[9]刘韩,吴丽云,高贺,等.易错PCR法提高土芽孢杆菌ZH1羧酸酯酶的热稳定性[J].微生物学报,2015,55(8):1 060-1 067.
[10]D EStephens,SSingh,K Permaul K.Error-Prone PCR of a Fungal Xylanase for Improvement of Its Alkaline and Thermal Stability[J].FEMS Microbiol Lett,2009,293(1):42-47.
[11]Y Jiguo,W Yonghua,Y Bo,et al.Degummol/Ling of Vegetable Oil by a New Microbial Lipase[J].Food Technol Biotech,2006,44(1):101-104.
[12]陈环,薛正莲,王洲,等.粘质沙雷氏菌PL-06磷脂酶A1在大肠杆菌中的分泌表达及其条件优化[J].食品工业科技,2015,36(16):193-197.
[13]高义平,赵和,吕孟雨,等.易错PCR研究进展及应用[J].核农学报,2013,27(5):607-612.
[14]黄瑛,蔡勇,杨江科,等.基于易错PCR技术的短小芽孢杆菌YZ02脂肪酶基因Bp L的定向进化[J].生物工程学报,2008,24(3):445-451.
Improving the Activity of Phospholipase A1 from Serratia Marcescs PL-06 by Error-prone PCR
WANG Yu-man,XUE Zheng-lian∗,WANG Zhou,SU Yan-nan,ZHU Hao
(College of Biological and Chemical Engineering,Anhui Polytechnic University,Wuhu 241000,China)
In order to obtain phospholipase A1 with higher activity,error-prone PCR was used to evolve the phospholipase A1 from Serratia marcescens PL-06 by adding different concentration of Mg2+and Mn2+.The gene was cloned into p ET28a(+)expression vector,and a mutant library was constructed.After the first and secondary screening,one mutant showed significant elevated activity,named EPB8.The enzyme activity of the mutants was 22.5%higher than that of the wild-type control.And increased by 41%after optimal Fermentation conditions.The sequence of the plaB gene of the mutants showed 7 mutated amino acids.It was found that phospholipase A1 protein revealed three amino acid substitutions:Y97C,P98S,X228L,and auxiliary protein revealed four amino acid substitutions:P46L,Q58L,N136D,H233R.These results provide a basis for further research of phospholipase A1.
Serratia marcescens;phospholipase A1;error-prone PCR;optimization of fermentation conditions
Q786
A
1672-2477(2016)04-0006-06
2016-01-10
国家自然科学基金资助项目(31471615)
王玉满(1989-),女,山东成武人,硕士研究生.
薛正莲(1967-),女,安徽和县人,教授,硕导.
猜你喜欢
中学生数理化·七年级数学人教版(2022年6期)2022-06-05
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
西南农业学报(2021年4期)2021-05-25
昆明医科大学学报(2021年2期)2021-03-29
中学生数理化(高中版.高考数学)(2021年12期)2021-03-08
中学生数理化(高中版.高考数学)(2020年10期)2020-10-27
山东农业科学(2017年8期)2017-09-09
天津医科大学学报(2015年2期)2015-12-22
江苏农业科学(2015年8期)2015-09-10
现代检验医学杂志(2015年2期)2015-02-06
