
子宫腺肌病组织中MMP2、MMP7、RECK、TWIST1、CD133表达及MVD变化
2016-09-06 07:19王木森柳雅玲
山东医药 2016年26期
王木森,柳雅玲
(1东阿县人民医院,山东东阿252200;2泰山医学院)
子宫腺肌病组织中MMP2、MMP7、RECK、TWIST1、CD133表达及MVD变化
王木森1,柳雅玲2
(1东阿县人民医院,山东东阿252200;2泰山医学院)
目的观察子宫腺肌病组织中基质金属蛋白酶2(MMP2)、基质金属蛋白酶7(MMP7)、反转录富含半胱氨酸蛋白(RECK)、TWIST1、CD133表达及微血管密度(MVD)变化,并探讨其意义。方法用免疫组化SP 法检测40例子宫腺肌病患者(观察组)在位、异位子宫内膜组织和40例子宫肌瘤患者(对照组)子宫内膜组织中的MMP2、MMP7、RECK、TWIST1、CD133、CD31,计算MMP2、MMP7、RECK、TWIST1、CD133染色指数,根据CD31染色结果计算MVD。结果MMP2、MMP7、TWIST1、CD133、MVD观察组异位内膜高于在位内膜,观察组在位内膜高于对照组子宫内膜(P均<0.05);RECK观察组异位内膜低于在位内膜,观察组在位内膜低于对照组子宫内膜(P均<0.05)。结论子宫腺肌病患者在位、异位子宫内膜组织中存在MMP-2、MMP7、TWIST1、CD133过表达、MVD升高及RECK蛋白表达缺失,且异位内膜甚于在位内膜。这些变化可能与子宫腺肌病的发病有关。
子宫腺肌病;上皮间质转化;异位内膜;在位内膜;基质金属蛋白酶
子宫腺肌病是妇科常见疾病,常表现为痛经、经期延长及不孕等,对妇女健康危害较大。其发病可能与免疫、血管生成、激素、细胞凋亡、细胞侵袭黏附能力及遗传等因素有关,确切机制至今尚未阐明[3]。2012年10月~2014年10月,我们检测了子宫腺肌病患者异位、在位子宫内膜组织中基质金属蛋白酶2(MMP2)、基质金属蛋白酶7(MMP7)、反转录富含半胱氨酸蛋白(RECK)、TWIST1、CD133蛋白表达及微血管密度(MVD)变化,探讨其与子宫腺肌病发病的相关性。
1 资料与方法
1.1临床资料选择东阿县人民医院病理科同期接收的40例术后诊断为子宫腺肌病的全子宫切除子宫组织标本为观察组,患者年龄25~45岁,平均36岁;同期40例术后诊断为子宫肌瘤的全子宫切除子宫组织标本为对照组,患者年龄22~43岁,平均35岁。两组子宫内膜均为增殖期,排除子宫内膜肿瘤性病变、卵巢恶性肿瘤及合并其他严重全身性疾病者,术前3个月均未服用过雌、孕激素类制剂治疗。
1.2MMP2、MMP7、RECK、TWIST1、CD133表达及MVD检测方法标本均于离体30 min内送病理科取材,4%甲醛固定4~6 h,脱水,石蜡包埋,连续4 μm切片。采用免疫组化SP法行MMP2、MMP7、RECK、TWIST1、CD133、CD31染色,操作按试剂盒说明书进行。采用Weidner计数法[5]计数CD31标记的微血管,选取5个高倍镜视野取其平均值即为MVD。其余蛋白结果判定采用Mattern积分法[4],染色指数=阳性率积分+染色强度积分。阳性率积分:阳性率≤25%为0分,阳性率>25%~≤50%为1分,阳性率>50%~≤75%为2分,阳性率>75%为3分;染色强度评分:细胞无着色为0分,染成细微的浅黄色颗粒为1分,较粗均匀的棕黄色颗粒为2分,较大深黄色颗粒为3分。

2 结果
MMP-2、MMP-7在内膜腺体及间质细胞细胞质表达,为黄色或淡棕色颗粒。RECK、TWIST1在内膜腺体细胞质表达,为黄色或淡黄色颗粒。CD133在子宫内膜腺体细胞细胞细胞质中表达,腺体腔缘着色强。MMP2、MMP7、TWIST1、CD133、RECK染色指数秩和见表1。由表1可见,观察组异位内膜MMP2、MMP7、TWIST1、CD133染色指数秩和高于在位内膜,观察组在位内膜MMP2、MMP7、TWIST1、CD133染色指数秩和高于对照组子宫内膜(P均<0.05);RECK染色指数秩和观察组异位内膜低于在位内膜,观察组在位内膜低于对照组子宫内膜(P均<0.05)。观察组异位内膜、在位内膜和对照组CD31所标记的MVD分别为(32.850±3.813)、(22.275±3.789)、(14.275±3.679)条/HP,观察组异位内膜高于观察组在位内膜,观察组在位内膜高于对照组(P均<0.05)。
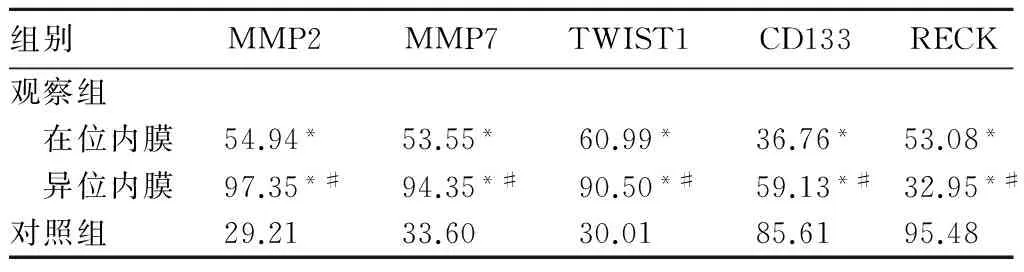
表1 两组MMP2、MMP7、TWIST1、CD133、RECK染色指数秩和比较
注:与对照组相比,*P<0.05;与在位内膜相比,#P<0.05。
3 讨论
子宫腺肌病表现为子宫内膜腺体和间质细胞向肌层内的浸润性生长,其分子机制大部分是未知的,是一个非常复杂的过程[6],至今未被完全阐明。免疫学变化、激素水平、微环境血管生成、遗传因素和表观遗传修饰等因素可能参与其中[7,8]。子宫腺肌病异位内膜腺体和间质细胞的侵袭性生长与基底膜及细胞外基质在周围微环境中的降解和重构有关系,并且与干细胞分化和上皮间质转化有关。
本研究结果显示,子宫腺肌病患者异位内膜MMP2、MMP7过表达,RECK表达缺失,可能与细胞外基质和基底膜的过度降解有关。这可能是子宫腺肌病有与恶性肿瘤类似的生物学行为的原因之一,即子宫内膜腺体和间质细胞异位到子宫肌层,并在肌层内侵袭性生长。有研究者在动物实验中发现,通过抑制MMPs的分泌,可阻止子宫内膜异位病灶的形成,也间接证实了MMPs在子宫腺肌病发生发展过程中发挥重要作用。MMP2还具有明胶结合区域,该区域由58个氨基酸残基组成的,属于与纤维连接素类似的明胶酶类,主要作用底物为Ⅳ型胶原蛋白; 活化的MMP2定位于细胞表面穿透细胞外基质的伸出部位,推测其在降解基底膜及细胞外基质成分Ⅳ型胶原过程中起着类似钻头作用。MMP7作为MMPs家族成员之一,同样拥有酶解基底膜和细胞外基质的能力。有研究提示,MMP7可以催化C型凝集素域家族3成员A酶解,导致细胞失去细胞间相互黏附的能力[9]。由于MMP7拥有比其他MMPs成员更广泛的底物范围,具备可以降解几乎所有细胞外基质的能力,例如Ⅲ型胶原、Ⅳ型胶原、蛋白聚糖、层粘连蛋白等。研究人员推测,这种特性可能与其简单的结构区域有关,导致缺少能够特异性识别底物的结构部分,从而具有更强的降解基质的能力。MMP2降解了Ⅳ型胶原和明胶,而MMP7降解了Ⅰ、Ⅲ、Ⅳ、Ⅴ型胶原,从而使细胞外基质和子宫肌层平滑肌纤维降解,导致异位内膜腺体和间质细胞在肌层内侵袭性生长。MMP2、MMP7的过度表达使得异位内膜的侵袭能力更强,相比于在位内膜,侵袭能力更强的异位内膜组织能降解子宫肌层中平滑肌细胞束周围的细胞外基质和基底膜,破坏了阻挡子宫内膜侵袭的固有屏障,为子宫腺肌病异位病灶的形成提供了方便条件[10]。MMPs蛋白高表达将导致细胞外基质过度降解,毛细血管及周围组织完整性变低,容易造成新生血管的形成,RECK蛋白的过高表达则抑制新生毛细血管的形成。
在胚胎分化发育过程中RECK基因起重要作用,包括在胚胎毛细血管的形成、神经肌肉系统发育、骨与软骨系统形成及神经肌肉连接处组织的分化、发育成熟过程中起着关键作用。作为新近发现的一种MMPs抑制剂,RECK可以抑制MMP2、MMP7及MT1-MMP的表达,从而达到抑制肿瘤的血管生成及浸润、转移的目的[11,12]。MMP7前体的分泌受到RECK基因调控抑制,而细胞膜表面锚定的RECK蛋白及可溶性RECK蛋白均可降低MMP2,MMP7及MT1-MMP的表达。其次,RECK蛋白可降低MMP2前体蛋白的转录活化水平,通过下调MT1-MMP的活性抑制MMP2前体蛋白向MMP2蛋白的转化[13]。
本研究结果显示,观察组异位内膜TWIST1、CD133表达高于在位内膜,在位内膜高于对照组,异位内膜中表达高于对照组内膜。TWIST1可能是通过促进腺上皮细胞向间质细胞转化,使其获得更强的侵袭性,从而在子宫腺肌病异位内膜的生长、蔓延中发挥着至关重要的作用。CD133蛋白选择性表达在胚胎肝脏、人骨髓、脐带血等造血细胞中[14,15],也表达在在一些干细胞表面。CD133表达随着细胞的逐渐分化而下调[16]。故国内外众多研究人员都把CD133作为肿瘤干细胞的标记分子。肿瘤细胞CD133高表达与其发生、侵袭、转移、耐药及复发有着密不可分的联系[17]。上皮细胞间质细胞转化是一个上皮细胞向间质细胞分化方向转化从而形成间质细胞的过程。这是一个可逆性和暂时性的过程,在这个过程中,上皮性标志物表达降低,间质细胞标志物表达升高,细胞获得了更强的侵袭能力,细胞蛋白质分子的表达也发生了改变。本研究结果提示,子宫腺肌病患者异位子宫内膜在分化程度和侵袭能力上更类似肿瘤组织。
子宫腺肌病患者异位子宫内膜的侵袭性生长离不开血液供应,因此研究人员推测微血管生成是子宫腺肌病的关键发病机制之一。国内杨尧华等[2]研究显示,血管生成因素可能在子宫腺肌病发病过程中发挥着不可忽视的作用。Hirotaka等[18]研究结果显示,接近50%的子宫腺肌病患者子宫内膜间质内出现过多的血管形成。本研究结果显示,观察组异位、在位子宫内膜与对照子宫内膜组织中均可见CD31阳性表达细胞,阳性染色表现为细小棕黄色颗粒,主要表达于子宫内膜间质血管内皮细胞膜或细胞质内。观察组异位内膜MVD显著高于在位内膜,在位内膜高于对照组。提示子宫腺肌病患者异位内膜与在位内膜具有显著增强的血管生成能力,且子宫内膜异位至肌层后其血管生成能力较在位子宫内膜显著增强,并强于对照组子宫内膜,更进一步显示子宫腺肌病患者异位内膜中微血管处于极其过度增生的活跃状态,以利于异位内膜侵袭生长,促使异位病灶进一步增大。
[1] 白红,赵丽嫣.子宫腺肌病病因与发病机制研究进展[J].中国妇产科临床杂志,2003,4(6):460-463.
[2] 杨尧华,周新艳,王岚,等.子宫腺肌病诊断及治疗的研究新进展[J].中国妇幼保健,2010,27(5):3992-3995.
[3] Nisolle M, Alvarez M, Colombo M, et al. Pathogenesis of endometriosis[J]. Gynecol Obstet Ferti, 2007,35(2):898-903.
[4] 杨钊. 胃癌血管生成拟态的初步研究[D].衡阳:南华大学,2011:253.
[5] Weidner N, Semple JP, Welch WR, et al. Tumor angiogenesisand metastasis: correlation in invasive breast carcinoma[J]. N Engl Med, 1991,324(1):1-8.
[6] 代玮,韩英莲,李荷莲.基质金属蛋白酶抑制剂RECK在子宫内膜异位症中的表达及临床意义[J].中国妇幼保健,2009,24(3):354-356.
[7] Jodele S, Blavier L, Yoon JM, et al. Modifying the soil to affect the seed: role of stromal-derived matrix metalloproteinases in cancer progression[J].Cancer Metastasis Rev, 2006,25(1):35-43.
[8] Rundhaug JE. Matrix metalloproteinases and angiogenesis[J].Cell Mol Med, 2005,9(2):267-285.
[9] Nisolle M, Alvarez M, Colombo M, et al. Pathogenesis of endometriosis[J]. Gynecol Obstet Ferti, 2007,35(3):898-903.
[10] May K, Becker CM. Endometriosis and angiogenesis[J]. Minerva Ginecol, 2008,60(3):245-254.
[11] 刘倩, 吴小翎, 罗红春. MMP-2 在胃癌组织中的表达及其与微血管生成和淋巴结转移的关系[J], 重庆医学,2012,41(13):1262-1267.
[12] Rundhaug JE. Matrix metalloproteinases and angiogenesis[J]. J Cell Mol Med, 2005,9(2):267-285.
[13] Loayza-Puch F, Yoshida Y, Matsuzaki T, et al. Hypoxia andRAS-signaling pathways converge on, and cooperatively downregulate,the RECK tumor-suppressor protein through microRNAs[J]. Oncogene, 2010,29(18):2638-2648.
[14] 唐毕锋,马立业,翟羽佳,等. 肿瘤干细胞标志物CD133在胃癌中的表达及其临床意义[J]. 临床肿瘤学杂志,2008,25(6):495-498.
[15] Chiao MT, Yang YC, Cheng WY, et al. CD133+glioblastoma stem-like cells induce vascular mimicry in vivo[J].Curr Neurovasc Res, 2011,8(3):210-219.
[16] Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells[J]. Cell, 2008,133(4):704-715.
[17] Yu Z, Pestell TG, Lisanti MP, et al. Cancer stem cells[J]. Int J Biochem Cell Biol, 2012, 44(12):2144-2151.
[18] Hirotaka M, Tetsuo M, Emi H, et al. Noninvasive and real time assessment of reconstructed functional human endometrium in NOD /SCID /Ccnullimmunodeficient mice[J]. PNAS, 2007,10(4):1925-1930.
柳雅玲(E-mail: tyylliu@sina.com)
10.3969/j.issn.1002-266X.2016.26.017
R71
B
1002-266X(2016)26-0053-03
2016-04-12)
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
北方牧业(2021年5期)2021-11-29
感染、炎症、修复(2021年1期)2021-07-28
中国民间疗法(2020年22期)2021-01-07
家禽科学(2020年1期)2020-12-18
世界科学技术-中医药现代化(2020年2期)2020-07-25
解放军医学院学报(2020年12期)2020-03-29
中成药(2019年12期)2020-01-04
中国卫生标准管理(2015年7期)2016-01-15
中国卫生标准管理(2015年13期)2016-01-15
