
基于正负样例的蛋白质功能预测
2016-08-31 03:49:38傅广垣余国先郭茂祖
计算机研究与发展 2016年8期
傅广垣 余国先 王 峻 郭茂祖
1(西南大学计算机与信息科学学院 重庆 400715)2 (哈尔滨工业大学计算机科学与技术学院 哈尔滨 150001)
基于正负样例的蛋白质功能预测
傅广垣1余国先1王峻1郭茂祖2
1(西南大学计算机与信息科学学院重庆400715)2(哈尔滨工业大学计算机科学与技术学院哈尔滨150001)
(gxyu@swu.edu.cn)
蛋白质功能预测是后基因组时代生物信息学的核心问题之一.蛋白质功能标记数据库通常仅提供蛋白质具有某个功能(正样例)的信息,极少提供蛋白质不具有某个功能(负样例)的信息.当前的蛋白质功能预测方法通常仅利用蛋白质正样例,极少关注量少但富含信息的蛋白质负样例.为此,提出一种基于正负样例的蛋白质功能预测方法(protein function prediction using positive and negative examples, ProPN).ProPN首先通过构造一个有向符号混合图描述已知的蛋白质与功能标记的正负关联信息、蛋白质之间的互作信息和功能标记间的关联关系,再通过符号混合图上的标签传播算法预测蛋白质功能.在酵母菌、老鼠和人类蛋白质数据集上的实验表明,ProPN不仅在预测已知部分功能标记蛋白质的负样例任务上优于现有算法,在预测功能标记完全未知蛋白质的功能任务上也获得了较其他相关方法更高的精度.
蛋白质功能预测;正样例;负样例;符号混合图;标签传播
实验发现蛋白质之间通过互作完成具体的生物学功能,这些互作的蛋白质构成互作网络[2-3].基于蛋白质互作网(protein-protein interaction network, PPI)的功能预测方法将每个蛋白质看作一个网络节点,节点之间的边则描述蛋白质间的互作关系.Schwikowski等人[2]发现互作的蛋白质之间通常共享相同的功能,进而提出一种基于互作邻居投票的功能预测方法.Chua等人[4]综合利用蛋白质的直接互作和间接互作刻画互作网络的全局结构,提出一种功能相似性度量方法重新计算蛋白质之间的互作强度,再基于互作邻居加权投票预测蛋白质功能.Deng等人[5]假设一个蛋白质的功能取决于与它直接互作的蛋白质,而与其他蛋白质无关,进而提出一类基于随机Markov场的功能预测方法.Mostafavi等人[6]整合多源蛋白质互作网络为一个复合网络,再基于复合网络预测蛋白质功能.
一个蛋白质通常参与到多个不同的生命过程中,具有多个功能,因而一个蛋白质通常标注有多个功能标记,上述方法均把功能预测问题当作二分类任务来处理,忽略了标记间的相关性.多标记学习能够利用标记间的关联性进而提高多标记对象的分类精度,近几年被广泛应用于蛋白质功能预测中[7-10].蛋白质的功能标记与基因本体有着密切的联系.基因本体(gene pntology, GO)[11]作为基因及其产物(如蛋白质)功能标注的通用标准已被广泛采用,它通过一个有向无环图描述功能标记间的层次结构关系,图中每个节点对应一种标记.标记间存在一个称为True Path Rule规则,当已知蛋白质标注有某个功能时,该蛋白质也标注有该功能标记的祖先节点对应的功能,而当蛋白质不应该标注有某个功能时,蛋白质也不会标注该功能标记所有子孙节点的功能.由蛋白质功能标记间的层次结构关系和True Path Rule可知标记之间存在一定的关联信息,Pandey等人[12]证实标记间的关联性可以提高蛋白质功能预测的精度.Zhang等人[7]利用Jaccard系数计算成对功能标记间的关联关系,再把这种关联关系结合到一种半监督分类框架中[13],提出一种基于高斯随机场的蛋白质功能预测方法(Gaussian random field, GRF).Wang等人[8]利用余弦函数和格林函数分别对标记之间的相关性和蛋白质之间的互作强度分别进行度量,提出一种基于功能相关性的多标记学习方法(function correlated multi-label learning, FCML)预测蛋白质功能.Yu等人[14]采用有向的双关系图模型描述蛋白质之间的互作信息,蛋白质的功能标记信息和标记之间的关联关系,提出一种直推式多标记分类方法(transductive multi-label classi-fication, TMC)预测蛋白质功能.Chi等人[15]假定蛋白质的功能与蛋白质之间的特征相似度存在关联,提出了一种基于余弦迭代(Cosine iterative algorithm, CIA)的蛋白质功能预测方法,每一次迭代CIA都将候选标记集中置信度最高的功能标记增量标注到待预测的蛋白质上,再基于蛋白质更新的功能标记集合重新计算蛋白质之间的相似度,如此迭代直至算法收敛或达到指定的迭代次数.Yu等人[16-17]通过综合利用功能标记间的关联性和多个蛋白质互作网络,提出基于多核集成和多网络对准的功能预测方法.
上述这些方法并没有较好地处理蛋白质功能标记的不平衡和稀疏特性[18].截止2016-03-01,GO中已有超过50 000个功能标记,它们分布在3个不同分支:生物过程(biological process, BP)、细胞成分(cellular component, CC)和分子功能(molecular function, MF),每个分支均用一个有向无环图描述功能标记间的层次结构关系.这些功能标记分布并不均衡,仅有少量功能标记被标注到成百上千个蛋白质,大部分标记被标注到的蛋白质个数都少于30.此外,一个蛋白质通常仅被标注了GO中几个或者几十个功能标记.因此从如此大量的候选功能标记集合中准确预测蛋白质功能非常困难.
由于蛋白质功能标记的开放世界假设(open world assumption)[19-20]特性,蛋白质功能标记数据通常仅提供蛋白质具有某个功能信息(正样例),极少登记蛋白质不具有某个功能的信息(负样例),原因是测定蛋白质所具有的全部功能非常困难,生物学家通常更关注蛋白质具有的功能.因此,蛋白质与大量功能标记的正负关联性未知.当前的绝大部分蛋白质功能预测方法通常假设这些未知的关联为蛋白质的负样例,由于很多蛋白质已有的功能标记信息并不完整,存在缺失[18-23],这种假设降低了蛋白质功能预测的精度和覆盖度.
大部分基于机器学习的蛋白质功能预测算法都需要一定量的蛋白质正负样例作为训练样本,从而获得具有较好判别性的蛋白质功能预测模型.Zhao等人[24]借助正样例-无标记数据(positive-unlabeled data)[25]学习的思想,将标注有某个功能标记的蛋白质作为正样例,未标注该功能标记但标注有其他功能标记的蛋白质作为未标记数据,再在训练分类器的过程中从未标记数据中选择负样例,进而实现蛋白质功能预测.
一些学者近期的研究也证实了蛋白质负样例在蛋白质功能预测中的重要性.Yu等人[26]发现在预测蛋白质缺失标记的过程中结合设定的负样例,不仅可以减小预测问题的规模,还能极大地提高功能预测的精度.一些方法采用启发式技巧预选一部分负样例,再结合已有的正样例预测蛋白质功能,并指明负样例在提高功能预测精度方面发挥着重要作用.Guan等人[27]假定所有未标注有某个功能标记的蛋白质为该功能标记的负样例,Mostafavi等人[6,28]和Cesa-Bianchi等人[29]将标注了某个功能标记但未标注该标记的兄弟标记的蛋白质作为该标记的负样例.由于已有蛋白质的功能标记信息并不完整,蛋白质实际可能具有这些兄弟标记对应的功能,这些启发式方法很容易选择错误的负样例.Youngs等人[30]提出了一种参数化贝叶斯先验方法(ALBias)预测蛋白质的负样例,该方法首先基于已有的蛋白质功能标记信息,计算当已知一个功能标记被标注到一个蛋白质上时,其他功能标记被标注在该蛋白质上的经验条件概率,再结合一个蛋白质已有的功能标记集合汇总这些条件概率,选取概率最低的标记作为该蛋白质的负样例,实验证明这种方法选择的负样例可以提高功能预测精度.但是ALBias只利用了蛋白质功能标记数据库中提供的每个蛋白质的直接功能标记信息,并未考虑标记间的层次结构关系,而已有研究发现标记间的结构关系在蛋白质功能预测中发挥着重要作用[18,31-33].Youngs等人[34]提出2种负样例选择方法SNOB(select negative via observed bias)和NETL(negative examples via topic likelihood).SNOB利用标记间的层次结构关系将一个蛋白质的直接功能标记的祖先标记增补到该蛋白质上,在此基础上重新计算标记之间的条件概率,再利用类似ALBias的方式选择负样例.NETL方法将每个蛋白质看作一个文档,所有标注到蛋白质的功能标记看作文档中的单词,然后利用LDA(latent Dirichlet allocation)[35]进行负样例预测.
上述方法在预测负样例的过程中,均基于蛋白质已有的正样例和GO结构信息,并不能利用已有的少量的负样例和蛋白质的其他特征信息,如蛋白质之间的互作和氨基酸序列信息等.当前,蛋白质功能标记数据库中已累计登记了几千个蛋白质负样例,但数量远小于正样例的数量[36].在分析上述研究工作的基础上,本文提出一种基于正负样例的蛋白质功能预测方法(protein function prediction using positive and negative examples, ProPN).ProPN首先通过一个有向符号混合图描述蛋白质之间的互作,蛋白质已有的正负样例和标记间的关联关系,再利用符号混合图上的标签传播算法预测蛋白质功能.在3个不同物种(Yeast,Mouse和Human)蛋白质数据集上的实验表明,本文提出的ProPN不仅能较其他相关方法更准确地预测蛋白质的负样例,在蛋白质功能预测上也获得较其他对比算法更高的精度.
1 基于正负样例的蛋白质功能预测
1.1有向符号混合图
基于蛋白质互作网的功能预测方法通常利用图来描述蛋白质之间的互作信息,图中每个节点对应一个蛋白质,节点间的连线描述节点间的互作,这类方法再将蛋白质已有正样例作为随机游走算子进行标签传播,预测蛋白质功能.虽然这种图也可以进行蛋白质正负样例的传播预测,但它并不能描述和利用功能标记间的关联信息.Wang等人[37]和Yu等人[14]设计的双关系图模型虽然能够刻画蛋白质正样例和功能标记间的关联信息,但仍无法描述和利用蛋白质负样例信息.
为了充分描述和利用蛋白质互作信息、蛋白质正负样例信息和功能标记间的关联性,本文设计了一种如图1所示的有向符号混合图.
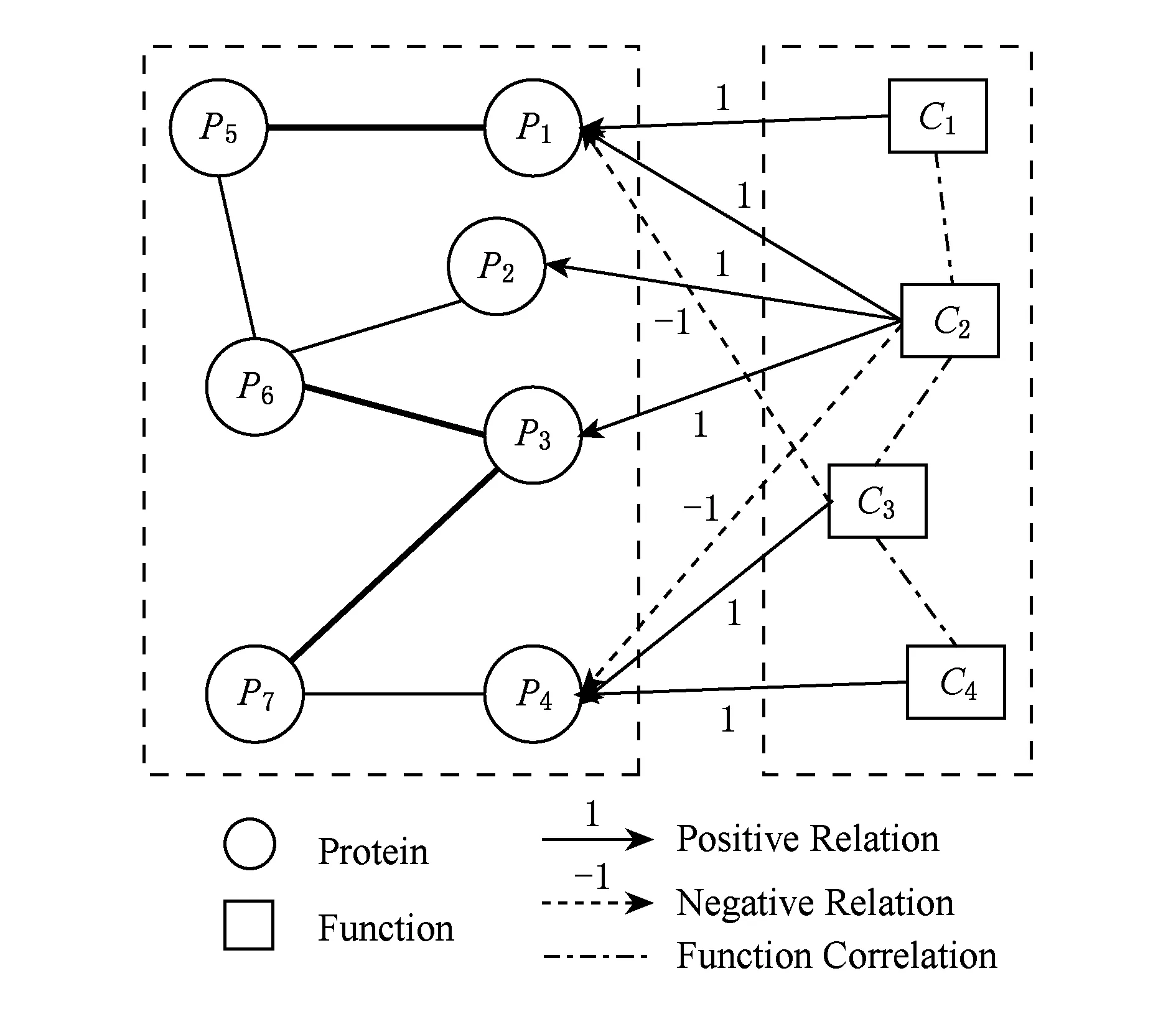
Fig. 1 Direct signed hybrid graph.图1 有向符号混合图示意图
图1中左侧子图中每个节点对应一个蛋白质,节点之间的连线描述蛋白质之间的互作,连线的粗细描述互作的强度(或置信度);右侧子图中每个节点对应一种功能标记,标记间的关联用虚线描述,线的粗细描述关联的大小;蛋白质与标记之间的正关联信息用有向实线(+1)描述,它们之间的负关联信息用有向虚线(-1)描述.为避免标签传播过程中标记被覆盖的风险,与文献[14]类似,本文的有向符号混合图仅允许功能标记节点上的信息传递到蛋白质节点.本文将通过有向混合图上的标签传播算法预测图中未完整标注功能蛋白质(如1图中P1,P2,P3和P4)的负样例和功能信息完全未知蛋白质(如图1中P5,P6和P7)的功能.值得注意的是本文的符号混合图中边的权重具有符号特性,而文献[14,37]的双关系图仅包含权重为非负的边,无符号特性.
1.2符号混合图上的标签传播算法
(1)
其中,WPP∈N×N描述蛋白质之间互作状况及其强度(或置信度),它由已有的蛋白质互作网络直接初始化;WFP∈C×N描述功能标记与蛋白质的关联关系,它可基于已有的蛋白质功能标记信息进行设置,当已知标记c为第i个蛋白质的正样例时WFP(c,i)=1,当已知c为第i个蛋白质的负样例时WFP(c,i)=-1,而当c与第i个蛋白质的关联信息未知时WFP(c,i)=0.由于图1为一个有向混合图,所以W的右上角的矩阵为一个大小为N×C的全0矩阵.WFF∈C×C描述功能标记间的关联关系,与文献[8,14]中的方法类似,本文基于成对功能标记共同标注在同一个蛋白质上的情况对其进行如下设置:
(2)
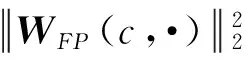
令Y=[Y1,Y2,…,YC],其中Yc∈N+C,描述第c个功能标记与N个蛋白质节点和C个功能节点的关联关系,具体定义如下:
(3)
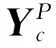
(4)
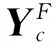
在上述初始化和设置的基础上,与文献[38]中局部与全局一致性学习的目标方程类似,本文定义图1中有向符号混合图上的标签传播目标方程为
(5)
f(i)∈C为预测的第i个节点与C个功能标记节点的关联性大小,Y(i)∈C表示第i个节点与C个功能标记节点的初始关联大小,D∈(N+C)×(N+C)为对角矩阵,其对角元素为,η>0用于调整式(5)中等号右侧第1项和第2项的权重.sgn(Wi j)为符号函数,当Wi j>0时取1,当Wi j<0时取-1.式(5)中等号右侧第1项为经验损失项,最小化该项的目的是促使预测的f(i)与已知的Y(i)保持一致.式(5)中右侧第2项为平滑性损失项,该项可细分为3种情况分别讨论:
1) 当i和j均为蛋白质节点时,Wi j≥0描述它们之间的互作强度(或氨基酸序列相似度等),当Wi j值较大时,最小化平滑损失项可以促使f(i)和f(j)与C个功能标记的关联性大小彼此靠近.
2) 当i为蛋白质节点而j为标记节点时,Wi j∈{-1,+1},其中Wi j=-1表示j为i的负样例,Wi j=1表示j为i的正样例,f(j)≥0描述标记j与C个标记节点的关联关系大小.若sgn(Wi j)=-1,最小化平滑损失项促使f(i)向-f(j)的靠近,而当sgn(Wi j)=1时会使f(i)向f(j)靠近,因此sgn(Wi j)可以保持并利用蛋白质正样例和负样例.
3) 当i和j均为功能标记节点时,Wi j≥0描述它们之间的关联大小,最小化平滑损失项可以促使f(i)与f(j)在C个功能标记节点上关联大小彼此靠近.当已知一个蛋白质与i正关联时,由于Wi j>0蛋白质也可能与j正关联;类似地,当一个蛋白质与i负关联时该蛋白质与j也可能负关联,这种设置有助于利用标记间的关联性.
综上所述,通过最小化式(5)可以预测已知部分功能标记蛋白质的负样例以及对完全未标注功能蛋白质的功能预测.
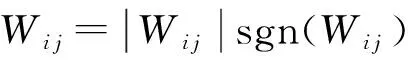
(6)
其中,tr()为求矩阵的迹,I是与S同型的单位矩阵,S是有向符号混合图W中各子图分别归一化的后关联矩阵,其定义如下:
(7)
其中:
(8)
(9)
(10)
DPP和DFF均为对角矩阵,它们的对角元素值分别为|WPP|和|WFF|行(或列)和.DP∈N×N和DF∈C×C也为对角矩阵,它们的对角元素值分别对应|WFP|的列和与行和.
对式(6)求关于f的导数可得:
(11)
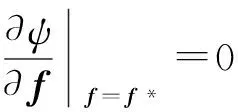
(12)
由此可得f*的显示解如下:
(13)
令α=1(1+η),则(1-α)=η(1+η),与文献[37]类似,式(11)可以转化为一个显式的迭代标签传播目标方程:
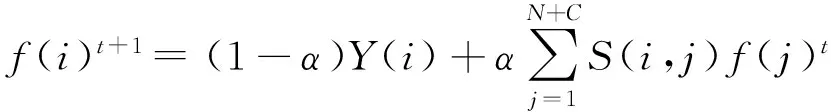
(14)
f(i)t∈C是第t次迭代时预测的第i个节点与C个功能标记的关联度大小,α用于平衡蛋白质i与功能标记的初始关联信息和其他节点传递给i的标记信息,α值越大说明其他节点传递的信息对f(i)影响越大.
令f0=Y,式(14)可以改写成如下的目标方程:
(15)
由于|αS(i,j)|<1,当t→+∞时式(15)中等号右侧第1项的极限为0、第2项为收敛的等比数列,其极限为(1-α)(I-αS)-1Y.在此基础上,可得式(14)的显式解如下:
f=(1-α)(I-αS)-1Y,
(16)
2 实 验
2.1数据集
本文下载了2016-02-21的GO数据库*http://geneontology.org,GO数据库描述了功能标记间的层次结构关系.这些标记分布在3个不相交的分支,分别是BP,MF和CC.与以往实验类似,本文剔除了GO中标注为“obsolete”的功能标记.本文采用Mostafavi等人[6]提供的Yeast,Human和Mouse蛋白质数据集,每个数据集中都包含了多个功能关联网络,本文将这些网络等权重合并为一个复合互作网络.本文同时也下载上述物种的功能标记数据*http://geneontology.org//page//download-annotations,这些标记数据提供了该物种当前已知的蛋白质的直接功能标记信息.为避免循环预测问题,本文剔除了证据属性(Evidence Code)为IEA(inferred from electronic annotation)的蛋白质功能标记.在此基础上,本文利用GO中的True Path Rule对蛋白质功能标记数据进行增补,将蛋白质正样例对应的标记节点的祖先节点标记也标注到相应的蛋白质上,蛋白质负样例对应标记节点的子孙节点也设定为相应蛋白质的负样例.
表1列出了上述3个数据集的统计信息,可以发现,蛋白质的正样例数量远大于其负样例的数量,少量的蛋白质直接负样例通过True Path Rule进行了一定的增补,每个物种的蛋白质均被上千个不同的功能标记所标注,其中Human在BP分支中的功能标记种类还大于其蛋白质数量.

Table1 Dataset Statistics表1 实验数据集统计信息
Note: Avg±Std is the average number of functions per protein and the standard deviation.
2.2对比方法与评价度量
本文实验分为2部分,预测已标注部分功能标记蛋白质的负样例和预测功能信息完全未知蛋白质的功能.
在负样例预测实验中本文选取当前最新的SNOB[34],ALBias[30]及基准方法Random作为ProPN的对比方法.Youngs等人实验验证SNOB比NETL效果更好[34],也优于启发式方法[6]和正样例无标记数据学习方法[24],所以本文没有采用这些方法作为对比方法.对于一个给定的功能c,ALBias对其作为蛋白质i负样例的关联性大小计算方式如下:
(17)
其中,Ai为蛋白质功能标记数据库中第i个蛋白质的直接功能标记集合,p(c|c′)表示经验条件概率:
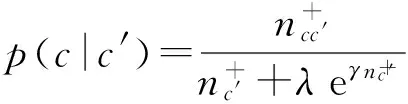
(18)

在对功能信息完全未知的蛋白质进行功能预测的实验中,本文选取CIA[15],FCML[8],GRF[7],TMC[14]和Naïve[1]作为ProPN的对比方法,前面4个方法在本文的引言中已经介绍,第5个方法Naïve是国际大规模蛋白质功能预测算法评测竞赛(critical assessment of protein function annotation algorithms, CAFA)中推荐的基准方法[1],Naïve基于功能标记出现在N个蛋白质上的频率进行预测,蛋白质标注某个功能标记的概率正比例于该标记的频率.在本部分实验中本文选取4个常用的评价度量MacroF1,MicroF1,Fmax和RankLoss评估上述蛋白质功能预测算法的性能.MicroF1将不同标记上的预测结果看作整体,求取全局的F1-Score作为评估结果,MicroF1更倾向频率高的标记.MacroF1是另外一种以标记为中心的评价度量,它求取每个功能标记的F1-Score,再基于这些标记F1-Score的均值评估预测性能,与MicroF1不同,MacroF1倾向频率低的标记.Fmax是CAFA推荐的一种以蛋白质为中心的评价准则[1],它基于全局准确率和查准率对应的F1分数的最大值评估预测性能. RankLoss是另外一种以蛋白质为中心的评价度量,它以每个蛋白质预测错误的功能标记排在正确标记前面的平均概率作为评价指标,显然RankLoss越低越好.为了保持与其他评价度量的一致性,本文实验中使用1-RankLoss作为评价度量,因此算法在上述4个度量上的值越高表明算法的预测性能越好.从这些评价度量的定义可知这些度量从不同的角度评估蛋白质功能预测的性能,一个算法很难在这些度量上均超过其他对比算法,关于这些度量的形式化定义可以参见文献[1,10,16].MicroF1和MacroF1需要将预测关联向量fP∈N×C转换成0-1的标记矩阵,与文献[7,14]类似,本文选取每个蛋白质对应预测关联向量中最大的K个元素对应的标记为该蛋白质的功能标记.K为数据集中每个蛋白质正样例的平均个数,例如在Human数据集的CC分支下,每个蛋白质平均标注有19.12个标记,则K=20.
2.3蛋白质负样例预测
ProPN利用2013年蛋白质功能标记数据设置蛋白质与功能标记之间的关联边,若已知一个蛋白质的正样例,则在该蛋白质与相关标记及其祖先标记之间设置权重为1的正关联边,若已知一个蛋白质负样例,则在该蛋白质与相关标记及其子孙标记之间设置权重为-1的负关联边.基于符号混合图上的标签传播算法得到N个蛋白质与C个功能标记的关联性大小矩阵fP∈N×C,参照SNOB和ALBias中的负样例选择与评价方法,本文通过对fP中全部N×C个元素按从小到大的顺序排序,选取前m个最小值对应的元素为N个蛋白质的负样例.然后基于2016年更新的蛋白质功能标记数据,统计m个预测的负样例中假阴性的数量FNs.对比算法ALBias,SNOB和Random同样也基于2013年的蛋白质功能标记数据进行负样例预测,再基于2016年的蛋白质功能标记数据统计各自产生的FNs.这些算法在不同的m下各自的FNs如表2~4所示:
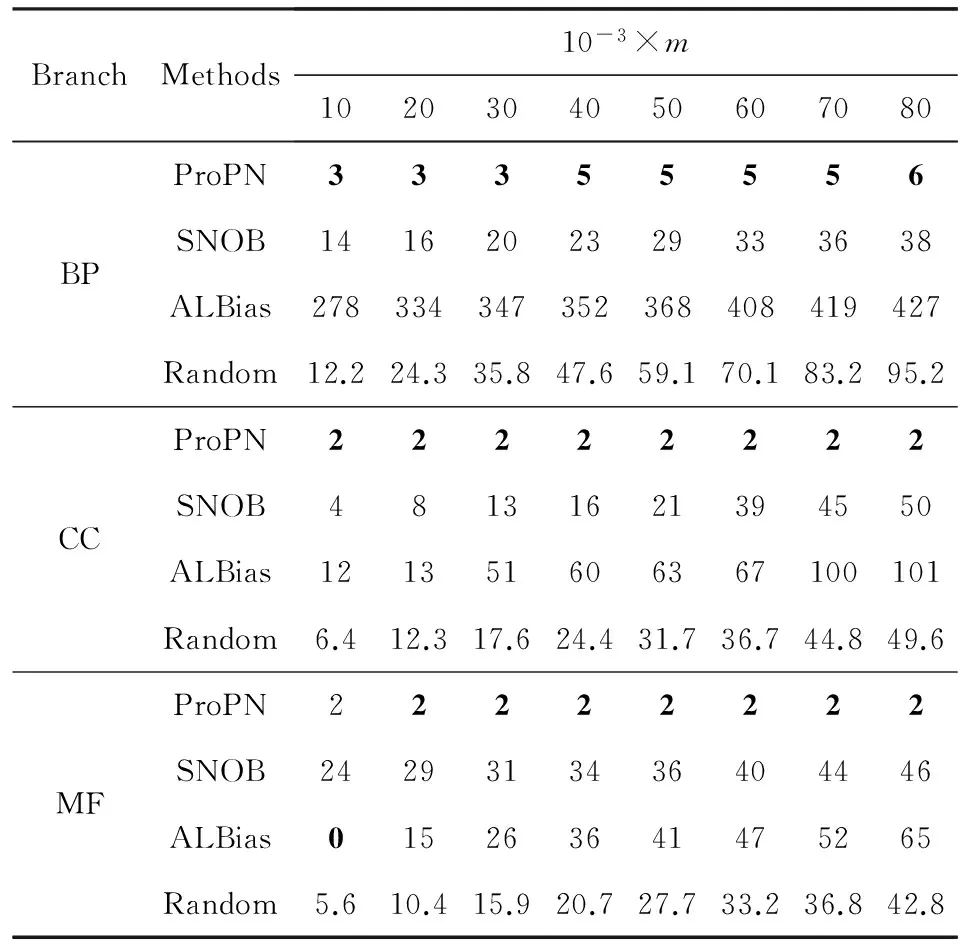
Table 2 Number of False Negative Predictions on Yeast Genome表2 Yeast数据集不同统计数量下的假阴性个数
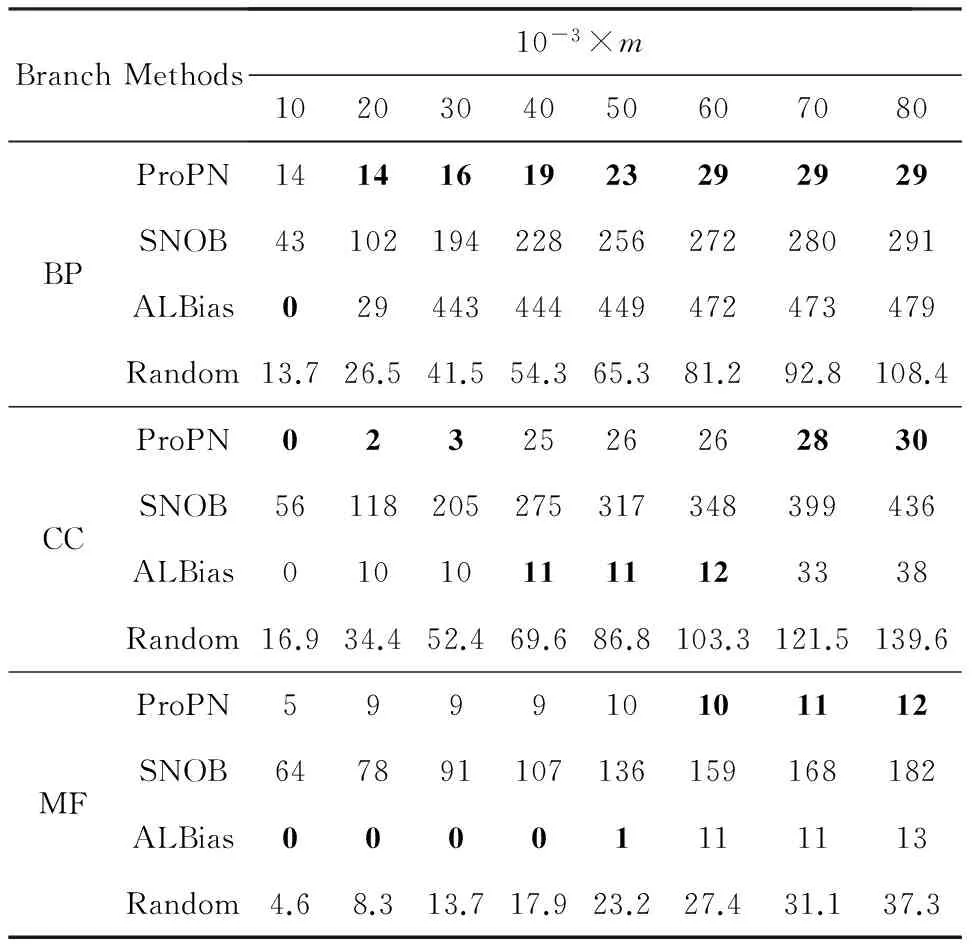
Table 3 Number of False Negative Predictions on Human Genome表3 Human数据集不同统计数量下的假阴性个数
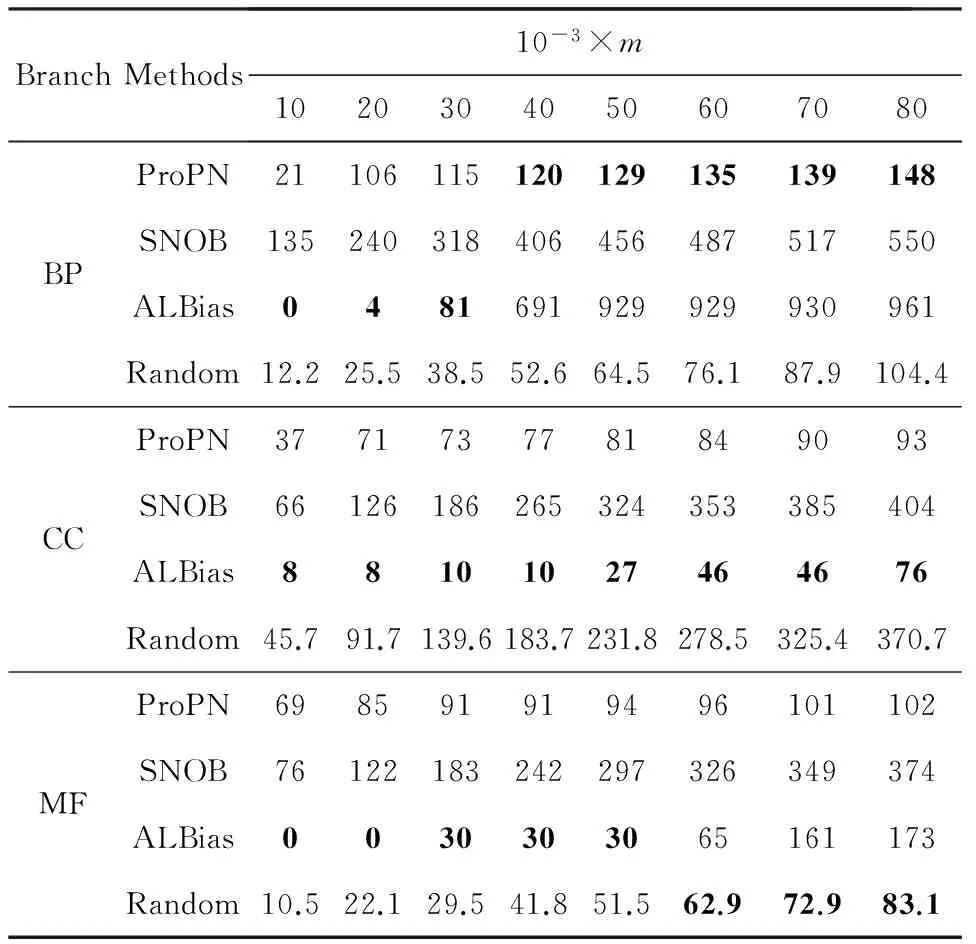
Table 4 Number of False Negative Predictions on Mouse Genome表4 Mouse数据集不同统计数量下的假阴性个数
从这3个表中的实验结果不难发现,本文提出的ProPN在绝大部分实验设置下都比其他对比算法精度高.以Yeast数据集BP分支上的实验为例,在m=80 k的情况下,ProPN有6个假阴性预测,SNOB有38个,ALBias有427个,而Random约有95.2个.上述实验结果证明了ProPN能够较当前负样例选择预测方法更准确地预测蛋白质的负样例.
SNOB通过True Path Rule补充了蛋白质的功能标记,在一定程度上利用标记间的层次结构关系,但它只在蛋白质正样例的基础上计算标记之间的经验条件概率,忽略了数量极少但信息丰富的蛋白质负样例,此外它没有考虑蛋白质之间的互作关系,因此它预测的FNs比ProPN多.ProPN同时考虑了蛋白质的正负样例和蛋白质的互作信息,预测的FNs较SNOB更少,这些实验结果表明结合蛋白质已有的负样例和其他特征信息进行负样例预测是有效的,也证明了利用有向符号混合图对上述信息进行描述和利用的必要性.
ALBias只利用了功能标记数据中提供的直接功能标记信息,对标记间层次结构关系的利用有限.与SNOB类似,ALBias也没有考虑蛋白质的互作信息和负样例,所以它在很多情况下预测的FNs比SNOB和ProPN多,这表明利用标记间的关系可以提高负样例的预测精度.ALBias在一些实验设置下较其他对比算法产生更少的FNs,原因是它通过λ和γ对蛋白质的缺失标记和标记不平衡进行了平滑处理.这说明在蛋白质的负样例预测任务中需考虑蛋白质功能标记信息的缺失情况,这也是本文后续研究工作之一.
Random仅基于标记出现的频率预测蛋白质的负样例,但它有时产生的FNs比SNOB和ALBias要少.原因是该方法并不是真正的随机选定功能标记为蛋白质的负样例,由于标记的层次结构特性,出现频率最低的标记最有可能预测为相关蛋白质的负样例,而这类负样例在新近的蛋白质功能标记数据中更难被验证.实际上ALBias和SNOB也偏向预测频率低的标记作为蛋白质负样例.
不同于这3个对比方法,ProPN通过有向符号混合图对蛋白质互作信息、已有蛋白质的正负样例和标记关联性进行描述和利用,在一定程度上避免了蛋白质功能标记的缺失性和不平衡性的影响,因此预测的负样例中FNs在绝大多数情况下均少于其他对比算法.
2.4蛋白质功能预测
在功能信息完全未知的蛋白质功能预测实验中,本文利用Yeast,Human和Mouse这3个物种蛋白质的2016年功能标记数据,随机将每个数据集划分为2部分,其中一部分蛋白质(占70%)被作为训练样本,剩余的蛋白质用作为测试样本.测试样本的功能标记在训练和预测过程中视为未知,仅在评测试阶段用于评估算法的性能.为避免随机性,本文对每个算法都独立重复10次,每次独立实验中均对训练样本和测试样本做一次随机划分,再报告这10次实验的平均结果和方差.Myers等人[39]认为标注到极少量蛋白质的功能标记无法在湿实验中测定,与其他对比方法[7,8,14]中的实验设置类似,本文过滤掉标注的蛋白质数量少于10的功能标记,过滤后3个数据集中每个蛋白质的功能标记情况见表1中最后一列.表5~7分别报告了各蛋白质功能预测算法在Yeast,Human和Mouse上的实验结果,3个表中加粗的实验结果为在95%置信度下成对t检验下最优的结果.在本部分实验中,为了提高有向符号混合图中节点间信息传播的速率,ProPN中的α和β分别设置为0.9和0.1,其他对比算法的参数设置采用原文作者推荐的方法进行设置.由于评价度量MacroF1和MicroF1需显式的将fP∈N×C转化为0-1矩阵,参照TMC和GRF等对比方法的实验方案,在蛋白质功能预测实验中,本文选取fP中每一行最大的K个元素对应的标记为该蛋白质的正样例,K为训练样本中每个蛋白质平均的正样例个数.1-RankLoss和Fmax不需要显示地将fP转化为0-1矩阵,它们直接基于fP进行计算.
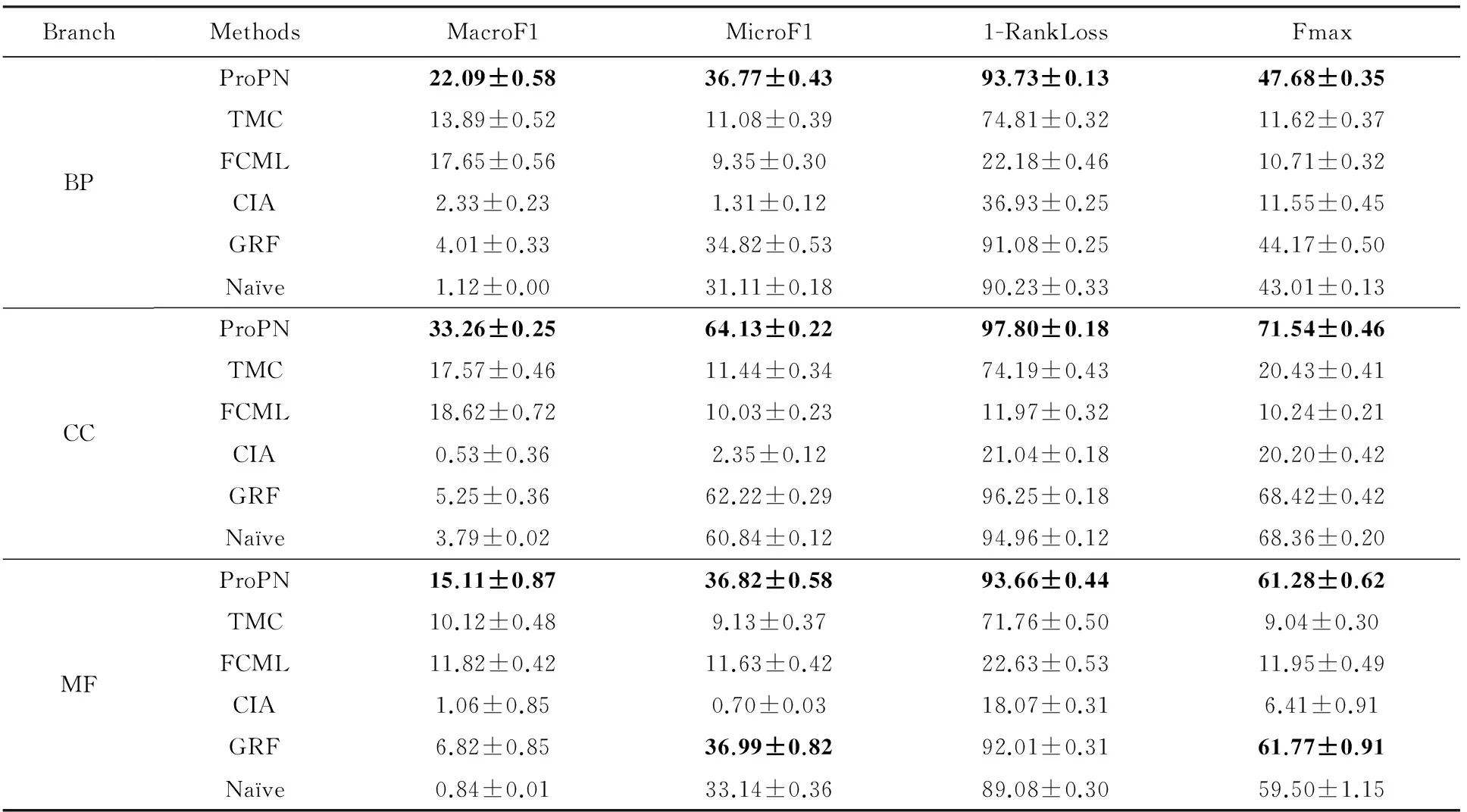
Table 5 Results on Yeast with Respect to Different Evaluation Metrics表5 Yeast数据集蛋白质功能预测评估结果
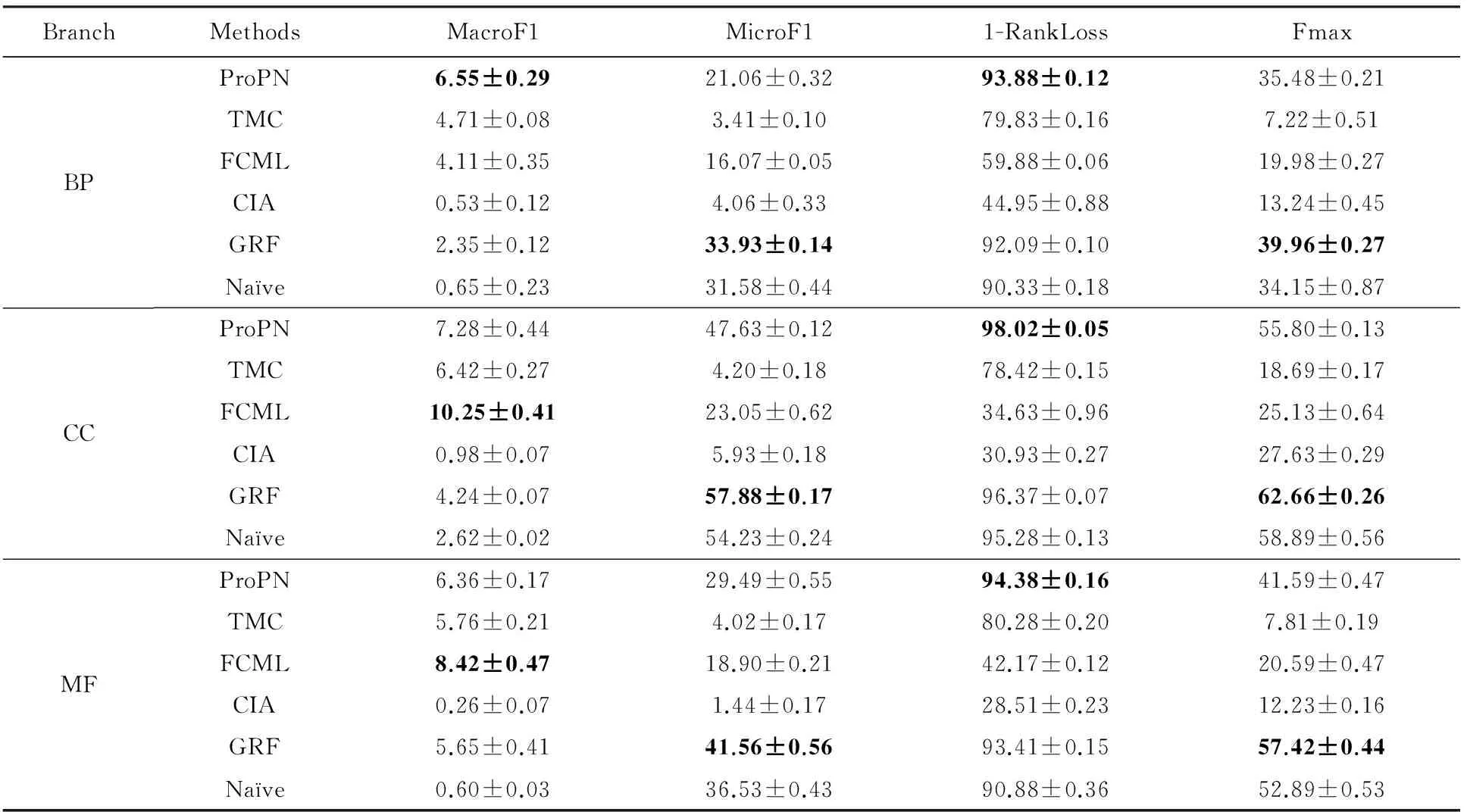
Table 6 Results on Human with Respect to Different Evaluation Metrics表6 Human数据集蛋白质功能预测评估结果
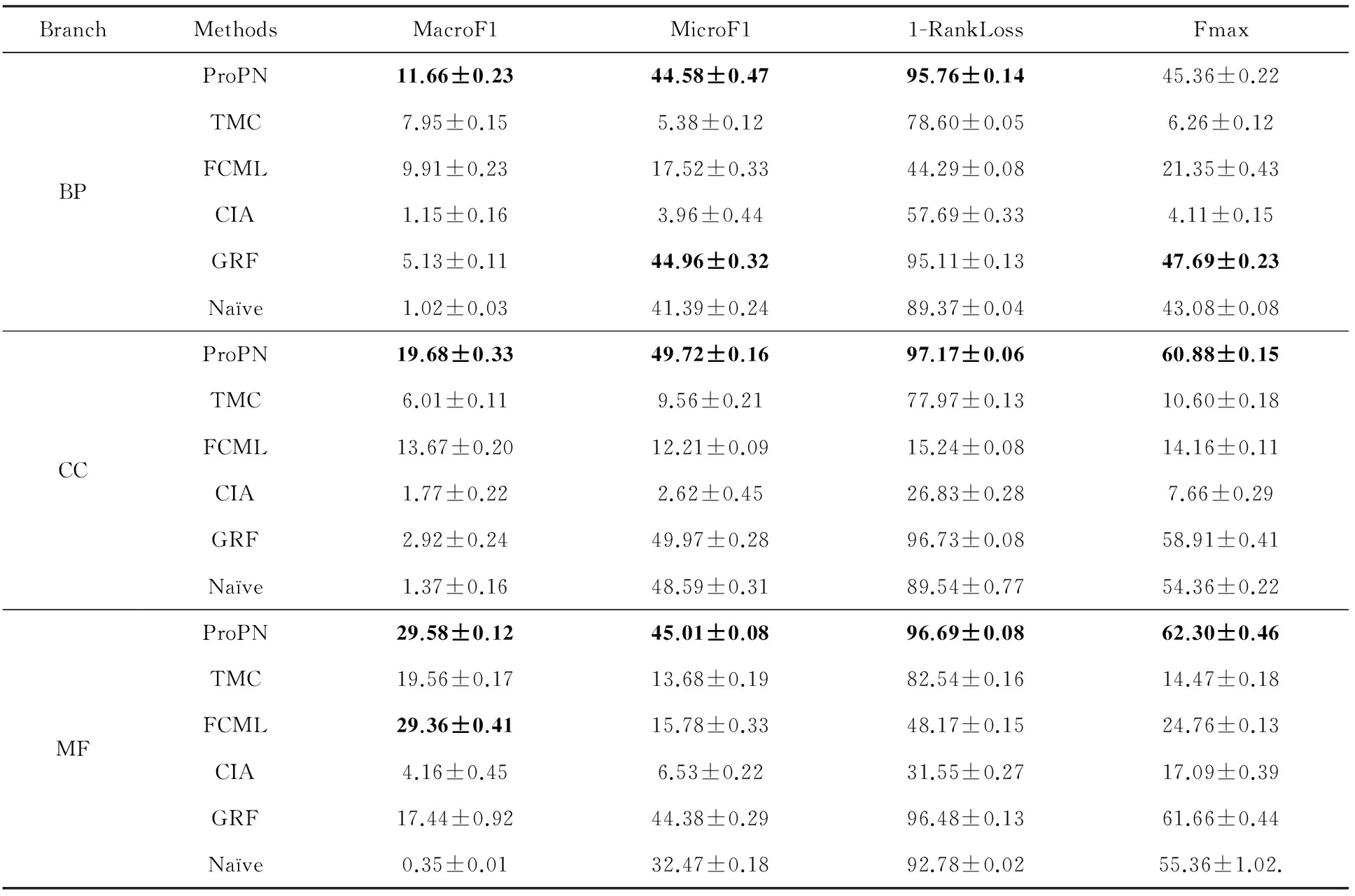
Table 7 Results on Mouse with Respect to Different Evaluation Metrics表7 Mouse数据集蛋白质功能预测评估结果
从表5~7中我们可以看到ProPN在绝大多数情况下均获得较这些对比算法更好的预测结果.具体地在本文36种(3种数据集×3个GO分支×4种评价度量)实验对比中,ProPN分别在72.2%, 86.1%和91.7%情况下显著性优于GRF,Naïve和FCML;ProPN在8.3%, 0%, 2.7%情况下获得与这3个对比方法类似的结果;ProPN在19.4%, 13.9%, 5.6%情况下比这3个方法差;ProPN总是获得较TMC和CIA更好的实验结果.
TMC和ProPN均能描述和利用蛋白质的互作信息,蛋白质与标记间的关联信息和标记间的关联关系,但ProPN的预测精度远高于TMC.原因是ProPN考虑了蛋白质的负样例并基于符号混合图对负样例信息进行了描述和利用,而TMC采用的有向双关系图无法对负样例进行描述和利用.ProPN与TMC预测结果的差异性也证实了负样例在蛋白质功能预测中的帮助作用.GRF与ProPN之间的预测结果差异最小,原因是GRF也利用了蛋白质之间的互作信息,标记间关联关系和与式(5)类似的目标方程预测蛋白质功能,但由于它没有利用蛋白质的负样例,所以它的精度通常低于ProPN.FCML也利用了标记之间的关联关系和蛋白质互作网,它有时获得与ProPN和GRF类似的结果,原因是它在蛋白质互作网对应的权重邻接矩阵上应用格林方程[8],在一定程度上降低了蛋白质噪声互作的干扰.这表明蛋白质之间互作信息的质量影响着基于互作网络的蛋白质功能预测算法的精度.虽然CIA也利用了蛋白质互作网进行功能预测,但它没有显式地考虑标记之间的关联性,因此其预测精度通常远低于上述对比算法.Naïve仅基于标记出现的频率预测蛋白质功能,它在4个评价度量上有时获得比其他对比算法更高的结果,特别是在MicroF1和1-RankLoss度量上,这种结果也与文献[1]中的结果类似.原因之一是功能标记的不平衡性和标记间的层次结构关系,高频率标记标注到蛋白质的概率更高,另一个原因是MicroF1更偏向频率高的标记.上述实验结果和分析证明本文提出的ProPN能够综合利用蛋白质的互作信息、标记间关联关系和蛋白质的正负样例,所以它获得较其他对比算法更高的预测结果.
2.5算法运行时间分析
为了分析各个对比算法的效率,与2.4节的实验设置类似,本文记录了5个对比方法(基线方法Naïve除外)各自的运行时间(算法评价度量除外),表8中报告的时间为算法5次独立运行的平均时间.这些方法均基于Matlab2011b(64 b)编码实现,实验运行平台配置为: Linux OS 3.13.0, AMD Opteron 6367, 64 GB RAM.
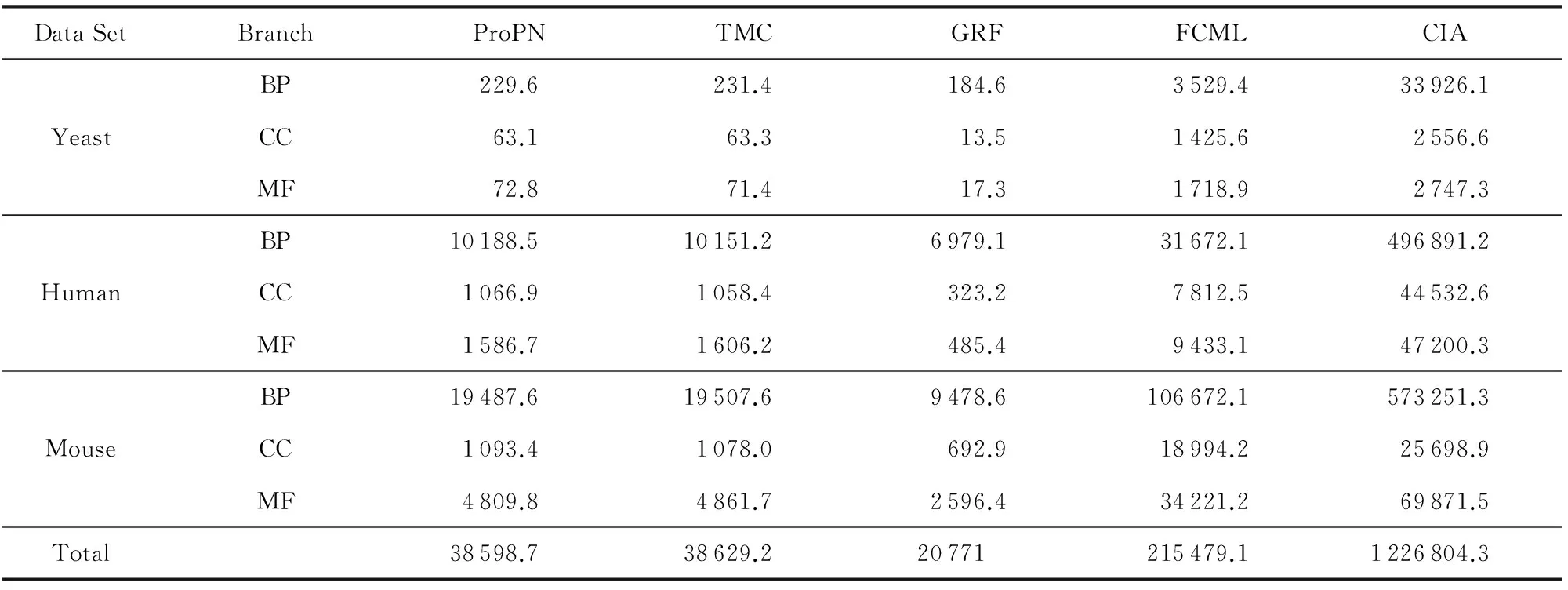
Table 8 Runtime Costs表8 运行时间 s
从式(12)可知图上标签传播算法的时间复杂度为O(N3),其中N为图中节点的个数,因此GRF和FCML的时间复杂度均为O(N3).由于ProPN和TMC采用的图中节点个数为N+C,所以其时间复杂度为O((N+C)3).CIA采用一种类似投票的机制预测蛋白质功能,但由于它需要多次迭代,每次迭代中都要重新计算N蛋白质之间的相似度大小和每个标记的局部和全局得分信息,因此其时间复杂度为O(tN2C),t为迭代次数.从表8也可以发现ProPN和TMC的时间耗费彼此接近,均大于GRF而又小于FCML和CIA,这是因为蛋白质互作网和蛋白质与功能标记的关联矩阵均为稀疏矩阵.FCML由于需要对蛋白质互作网对应的权重关联矩阵进行本征值分解,非常耗时,所以其真实时间耗费远大于其他算法,CIA由于需要多次迭代,每次迭代增量地更新蛋白质的功能标记集合和重新计算蛋白质之间的相似度,所以其时间耗费比较大.上述实验结果表明本文提出的ProPN不仅能较其他对比算法更准确地预测蛋白质功能,还能保持较高的效率.
3 结束语
本文将蛋白质的正负样例信息和蛋白质的互作信息综合应用于蛋白质功能预测当中,获得了较其他相关算法更好的预测效果,得到了若干有指导意义的结论.与现有蛋白质功能预测方法相比,本文不仅利用了蛋白质与功能标记的正关联信息,还利用了其中为数不多但富含信息的蛋白质与功能标记的负关联信息,为后续蛋白质功能预测方法的研究提供了新的思路.如何有效预测蛋白质负样例并结合负样例到蛋白质功能预测任务中、如何对蛋白质的潜在缺失标记进行建模均是值得深入研究的问题.此外,蛋白质互作网络质量和标记之间的关联性度量也影响着ProPN的性能,如何克服互作网中的噪声互作、如何更准确地描述功能标记间的关联关系都有待进一步研究.ProPN算法的matlab代码和部分采用的数据集可在www.scholat.comteammlda下载.
[1]Radivojac P, Clark W, Oron T, et al. A large-scale evaluation of computational protein function prediction[J]. Nature Methods, 2013, 10(3): 221-227
[2]Schwikowski B, Uetz P, Field S. A network of protein-protein interactions in yeast[J]. Nature Biotechnology, 2000, 18(12): 1257-1261
[3]Guo Maozu, Dai Qiguo, Xu Liqiu, et al. On protein complexes identifying algorithm based on the novel modularity function[J]. Journal of Computer Research and Development, 2014, 51(10):2178-2186 (in Chinese)
(郭茂祖, 代启国, 徐立秋, 等. 一种蛋白质复合体模块度函数及其识别算法[J]. 计算机研究与发展, 2014, 51(10): 2178-2186)
[4]Chua H, Sung W, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein-protein interactions[J]. Bioinformatics, 2006, 22(13): 1623-1630
[5]Deng M, Tu Z, Sun F, et al. Mapping Gene Ontology to proteins based on protein-protein interaction data[J]. Bioinformatics, 2004, 20(6): 895-902
[6]Mostafavi S, Morris O. Fast integration of heterogeneous data sources for predicting gene function with limited annotation[J]. Bioinformatics, 2010, 26(14): 1759-1765
[7]Zhang X, Dai D. A framework for incorporating functional interrelationships into protein function prediction algorithms[J]. IEEE//ACM Trans on Computational Biology and Bioinformatics, 2012, 9(3): 740-753
[8]Wang H, Huang H, Ding C. Function-function correlated multi-label protein function prediction over interaction networks[J]. Journal of Computational Biology, 2013, 20(4): 322-343
[9]Wu J, Huang S, Zhou Z. Genome-wide protein function prediction through multi-instance multi-label learning[J]. IEEE//ACM Trans on Computational Biology and Bioinformatics, 2014, 11(5): 891-902
[10]Zhang M, Zhou Z. A review on multi-label learning algorithms[J]. IEEE Trans on Knowledge & Data Engineering, 2014, 26(8): 1819-1837
[11]Ashburner M, Ball C A, Blake J A, et al. Gene ontology: tool for the unification of biology[J]. Nature Genetics, 2000, 25(1): 25-29
[12]Pandey G, Myers C, Kumar V. Incorporating functional inter-relationships into protein function prediction algorithms[J]. BMC Bioinformatics, 2009, 10: No.142
[13]Belkin M, Niyogi P, Sindhwani V. Manifold regularization: A geometric framework for learning from labeled and unlabeled examples[J]. Journal of Machine Learning Research, 2006, 7(1): 2399-2434
[14]Yu G, Domeniconi C, Rangwala H, et al. Transductive multi-label ensemble classification for protein function prediction[C] //Proc of 18th ACM SIGKDD Int Conf on Knowledge Discovery and Data Mining. New York: ACM, 2012: 1077-1085
[15]Chi X, Hou J. An iterative approach of protein function prediction[J]. BMC Bioinformatics, 2011, 12: No.437
[16]Yu G, Rangwala H, Domeniconi C, et al. Predicting protein function using multiple kernels[J]. IEEE//ACM Trans on Computational Biology and Bioinformatics, 2015, 12(1): 219-233
[17]Yu G, Zhu H, Domeniconi C, et al. Integrating multiple networks for protein function prediction[J]. BMC Systems Biology, 2015, 9(S1): S3
[18]Tao Y, Sam L, Li J, et al. Information theory applied to the sparse gene ontology annotation network to predict novel gene function[J]. Bioinformatics, 2007, 23(13): i529-i538
[19]Rhee S, Wood V, Dolinski K, et al. Use and misuse of the gene ontology annotations[J]. Nature Review Genetics, 2008, 9(7): 509-515
[20]Schnoes M, Ream D, Thorman A, et al. Biases in the experimental annotations of protein function and their effect on our understanding of protein function space[J]. PLoS Computational Biology, 2013, 9(5): No.e1003063
[21]Yu G, Zhu H, Domeniconi C, et al. Predicting protein function via downward random walks on a gene ontology[J]. BMC Bioinformatics, 2015, 16: No.273
[22]Legrain P, Aebersold R, Archakov A, et al. The human proteome project: current state and future direction[J]. Molecular and Cellular Proteomics, 2011, 10(7): M111.00999
[23]Gao Lei, Li Xia, Guo Zheng, et al. Broadly predicting specific protein functions with protein-protein interactions and gene expression profiles[J]. China Science: Life Science, 2006, 36(5): 441-450 (in Chinese)
(高磊, 李霞, 郭政,等. 结合蛋白质互作与基因表达谱信息大范围预测蛋白质的精细功能[J]. 中国科学: 生命科学, 2006, 36(5): 441-450)
[24]Zhao X, Wang Y, Chen L, et al. Gene function prediction using labeled and unlabeled data[J]. BMC Bioinformatics, 2008, 9: No.57
[25]Elkan C, Noto K. Learning classifiers from only positive and unlabeled data[C] //Proc of the 14th ACM SIGKDD Int Conf on Knowledge Discovery and Data Mining. New York: ACM, 2008: 213-220
[26]Yu G, Rangwala H, Domeniconi C, et al. Protein function prediction with incomplete annotations[J]. IEEE//ACM Trans on Computational Biology and Bioinformatics, 2014, 11(3): 579-591
[27]Guan Y, Myers C, Hess D, et al. Predicting gene function in a hierarchical context with an ensemble of classifiers[J]. Genome Biology, 2008, 9(S1): S3
[28]Mostafavi S, Ray D, Warde-Farley D, et al. GeneMANIA: A real-time multiple association network integration algorithm for predicting gene function[J]. Genome Biology, 2008, 9(S1): S4
[29]Cesa-Bianchi N, Re M, Valentini G. Synergy of multi-label hierarchical ensembles, data fusion, and cost-sensitive methods for gene functional inference[J]. Machine Learning, 2012, 88(1//2): 209-241
[30]Youngs N, Duncan P, Kevin D, et al. Parametric Bayesian priors and better choice of negative examples improve protein function prediction[J]. Bioinformatics, 2013, 29(9): 1190-1198
[31]Yu G, Zhu H, Domeniconi C. Predicting protein function using incomplete hierarchical labels[J]. BMC Bioinformatics, 2015, 16: No.1
[32]Pena-Castillo L, Tasan M, Myers C, et al. A critical assessment of mus musculus gene function prediction using integrated genomic evidence[J]. Genome Biology, 2008, 9(S1): S2
[33]Valentini G. True path rule hierarchical ensembles for genome-wide gene function prediction[J]. IEEE//ACM Trans on Computational Biology and Bioinformatics, 2011, 8(3): 832-547
[34]Youngs N, Penfold-Brown D, Bonneau R, et al. Negative example selection for protein function prediction: the NoGO database[J]. PLoS Computational Biology, 2014, 10(6): e1003644
[35]Blei D, Ng A, Jordan M. Latent dirichlet allocation[J]. Journal of Machine Learning Research, 2003, 3: 993-1022
[36]Huntley R, Sawford T, Martin M, et al. Understanding how and why the Gene Ontology and its annotations evolve: the GO within UniProt[J]. GigaScience, 2014, 3: No.4
[37]Wang H, Huang H, Ding C. Image annotation using bi-relational graph of images and semantic labels[C] //Proc of the 24th IEEE Conf on Computer Vision and Pattern Recognition. Piscataway, NJ: IEEE, 2011: 793-800
[38]Zhou D, Bousquet O, Lal T, et al. Learning with local and global consistency[C] //Advance in Neural Information Processing Systems. Cambridge, MA: MIT Press, 2003: 321-328
[39]Myers C, Barrett D, Hibbs M, et al. Finding function: evaluation methods for functional genomic data[J]. BMC Genomics, 2006, 7(1): No.187

Fu Guangyuan, born in 1993. Master. Student member of China Computer Federation. His main research interests include machine learning and bioinfor-matics.

Yu Guoxian, born in 1985. Associated professor. Member of China Computer Federation. His main research interests include machine learning, data mining and bioinformatics (gxyu@swu.edu.cn).

Wang Jun, born in 1983. Associated professor. Member of China Computer Federation. Her main research interests include data mining and bioinformatics (kingjun@swu.edu.cn).

Guo Maozu, born in 1966. Professor, PhD supervisor. Member of China Computer Federation. His main research interests include machine learning, data mining and bioinformatics (maozuguo@hit.edu.cn).
Protein Function Prediction Using Positive and Negative Examples
Fu Guangyuan1, Yu Guoxian1, Wang Jun1, and Guo Maozu2
1(CollegeofComputerandInformationScience,SouthwestUniversity,Chongqing400715)2(SchoolofComputerScienceandTechnology,HarbinInstituteofTechnology,Harbin150001)
Predicting protein function is one of the key challenges in the post genome era. Functional annotation databases of proteins mainly provide the knowledge of positive examples that proteins carrying out a given function, and rarely record the knowledge of negative examples that proteins not carrying out a given function. Current computational models almost only focus on utilizing the positive examples for function prediction and seldom pay attention to these scarce but informative negative examples. It is well recognized that both positive and negative examples should be used to achieve a discriminative predictor. Motivated by this recognition, in this paper, we propose a protein function prediction approach using positive and negative examples (ProPN) to bridge this gap. ProPN first utilizes a direct signed hybrid graph to describe the positive examples, negative examples, interactions between proteins and correlations between functions; and then it employs label propagation on the graph to predict protein function. The experimental results on several public available proteomic datasets demonstrate that ProPN not only makes better performance in predicting negative examples of proteins whose functional annotations are partially known than state-of-the-art algorithms, but also performs better than other related approaches in predicting functions of proteins whose functional annotations are completely unknown.
protein function prediction; positive examples; negative examples; signed hybrid graph; label propagation
2016-03-21;
2016-05-25
国家自然科学基金项目(61402378,61571163,61532014);重庆市基础与前沿研究项目(cstc2014jcyjA40031,cstc2016jcyjA0351);重庆市研究生科研创新项目(CYS16070);中央高校基本科研业务费基金项目(2362015XK07,XDJK2016B009,XDJK2016D021)
余国先(gxyu@swu.edu.cn)
TP391
This work was supported by the National Natural Science Foundation of China (61402378,61571163,61532014), the Natural Science Foundation of CQ CSTC (cstc2014jcyjA40031,cstc2016jcyjA0351), Chongqing Research Innovation Fund for Graduate (CYS16070), and the Fundamental Research Funds for the Central Universities of China (2362015XK07,XDJK2016B009,XDJK2016D021).
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12 02:07:42
肝博士(2022年3期)2022-06-30 02:48:48
海外星云(2021年9期)2021-10-14 07:26:10
国际眼科杂志(2021年9期)2021-09-15 03:24:42
装备制造技术(2020年2期)2020-12-14 03:09:16
当代陕西(2019年15期)2019-09-02 01:52:00
学苑创造·A版(2018年11期)2018-02-01 06:29:20
读者(2017年5期)2017-02-15 18:04:18
青苹果·教育研究版(2016年9期)2016-12-23 11:52:36
中国卫生(2015年12期)2015-11-10 05:13:34
