
芳族羧酸脱羧偶联反应的研究进展
2016-08-17 01:57李永凯谢媛媛浙江工业大学药学院杭州310014
浙江化工 2016年7期
李永凯,谢媛媛(浙江工业大学药学院,杭州 310014)
医药化工
芳族羧酸脱羧偶联反应的研究进展
李永凯,谢媛媛*
(浙江工业大学药学院,杭州310014)
含羧基的化合物可以通过脱羧偶联反应形成新的碳碳或碳杂键,该反应具有高度的选择性且主要副产物仅为二氧化碳。因此,通过脱羧偶联反应来构建化学键是一种廉价、环境友好的方法。对过去几年中芳香族羧酸脱羧偶联反应的研究进展做一简要综述。
芳香族羧酸;脱羧;偶联
0 前言
偶联反应在药物、天然生物活性产物和材料的合成中产生了深远的影响[1]。通常,这些反应涉及芳基或烷基卤化物与低价态金属氧化加成,再通过金属交换转移、还原消除得到预期产物[2]。在偶联反应中,过渡金属催化这一步通常使用相对昂贵或有毒的试剂。找到一条反应条件温和、绿色环保、使用廉价的载体合成有机金属中间体的方法是很有必要的。
随着药物合成的深入研究,发现脱羧偶联反应满足以上的需求。脱羧偶联反应利用金属脱羧形成有机金属中间体,最后通过还原消除发生偶合。与传统的交叉偶联方法相比,脱羧偶联具有几个方面优势:(1)羧酸衍生物具有储存方便、使用简单、结构稳定、价格便宜的特点;(2)脱羧可以在中性条件下进行,形成反应中间体;(3)反应的主要副产物是二氧化碳,绿色环保。此外,与通过碳氢活化得到反应活性中间体相比较而言,在特定位点通过脱羧反应更容易生成活性中间体[3]。
1 合成路线
芳香族类化合物是重要的药物中间体,合成芳香类化合物的方法很多,本文重点介绍的方法是通过脱羧偶联反应构建新的碳碳或碳杂键,使简单的芳香类化合物转变成重要的药物中间体。
1.1分子间脱羧偶联反应
1966年,Nilsson[4]就对脱羧偶联反应进行了尝试,发现邻硝基苯甲酸和当量的碘化亚铜,在高温下反应脱去羧基所形成的有机金属中间体,可以被捕获并进一步与碘代苯反应生成联苯(Scheme 1)。
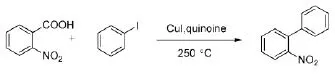
Scheme 1
但是该反应的反应条件极为苛刻,反应温度高达250℃,需要使用价格昂贵的碘代化合物作为反应物,并且产率低于20%,这些缺陷大大地限制了这类反应在有机合成中的应用。
2006年,Goossen发现了高产率的脱羧偶联方法。Goossen[5-6]及其合作者的设想是采用双金属催化剂体系:铜和钯共同作用来实现目标反应,如(Figure 1)所示。
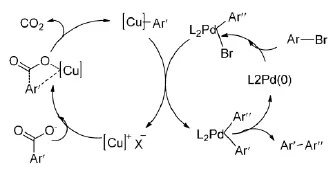
Figure 1
研究发现,以N-甲基吡咯烷酮(NMP)作为溶剂、Pd(acac)2作为催化剂、二苯基异丙基膦作为配体、氟化钾(KF)作为碱,使用1.5当量的碳酸铜可以在较低的温度下(120℃)得到相应的联苯化合物(Scheme 2)。

Scheme 2
该反应具有很好的化学选择性,仅得到很少量的脱羧自偶联和卤代烃自偶联副产物。反应体系中的质子氢对于反应产率有至关重要的影响,在反应体系中加入了适量的分子筛除去体系中的水可以有效地提高反应产率。邻位带有吸电子基团的芳香羧酸都能在该条件下顺利进行反应。
重新优化后的反应条件只需使用1 mol%的乙酰丙酮钯Pd(acac)2和3 mol%的CuI、5 mol% 的1,10-菲啰啉作为配体、碳酸钾作为碱。但是,为了提高产率,反应温度需要升高至160℃(Scheme 3)。由于该反应原料十分廉价并且只需使用催化量的过渡金属催化剂钯和铜,因此该反应具有很大的工业应用价值。
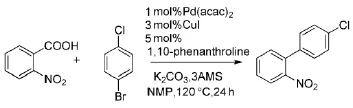
Scheme 3
进一步研究发现:在上述催化体系中加入少量的膦作为共同配体,可以使反应活性较低的氯代苯也能和羧酸发生脱羧偶联反应,收率高达88%[7]。氯代芳烃比溴代芳烃更稳定、廉价,使得该反应体系更加具有应用价值。此外,该反应体系还可以与三氟甲磺酸类化合物发生脱羧偶联反应,产率高达91%[8](Scheme 4)。
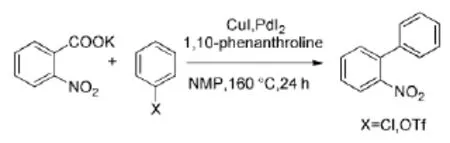
Scheme 4
Goossen[9]深入研究发现:在原有反应体系的基础上,把碘化亚铜替换为较为廉价的氧化亚铜,也能实现三氟甲磺酸类化合物与羧酸的偶联,收率高达91%(Scheme 5)。实验表明,邻位硝基苯甲酸的活性要比间位的活性高。然而,为了提高产率,反应温度需升高到170℃,反应条件较苛刻。
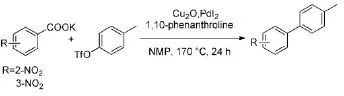
Scheme 5
Goossen等[10]人随后又发现,相类似的底物在碳酸银、醋酸铜(1.0 equiv)催化下,以DMF为反应溶剂,能与硅酸甲酯发生脱羧偶联,该反应的温度较低,为145℃,反应时间为18 h(Scheme 6)。该反应以氧气作为氧化剂参与反应,符合绿色环保的要求。该反应也为芳基上C-O键的构建提供了新的方法。

Scheme 6
该反应的机理为:底物A首先与银盐交换,生成中间体B,中间体B脱去二氧化碳,生成中间体C,随后与铜盐交换,生成中间体D,经过氧化形成三价有机铜E,中间体E与亲核试剂反应,通过还原消除生成产物F,如(Figure 2)所示。

Figure 2
通过实验证明该反应必须要有铜盐的参与,没有铜盐反应无法进行。同时也证明了反应机理的合理性,如(Figure 3)所示。
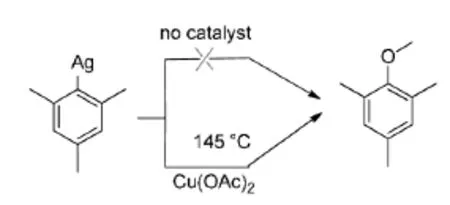
Figure 3
Crabtree等[11]发现:在微波反应器中使用醋酸钯作为催化剂,2,6-二甲氧基苯甲酸可以和苯甲醚发生脱羧偶联反应,得到联苯产物。该反应的特点是脱羧以后形成的有机金属中间体可以直接和C-H键反应生成新的联苯类化合物,将脱羧反应和C-H活化有机地结合起来(Scheme 7)。但是,该反应的缺点也十分明显:反应温度高达200℃,产率低于50%,并且反应区域选择性不好。

Scheme 7
赵品静等[12]人报道了利用相同的底物与丙烯酸叔丁酯在有机金属铑的催化下,生成重排的脱羧偶联产物。该反应使用了环辛-1,5-二烯铑,配体1,1-联萘-2,2-双二苯膦,在120℃,甲苯/水(5:1)的碱性条件下进行,收率达到71%。但反应时间太长,需要48 h(Scheme 8)。
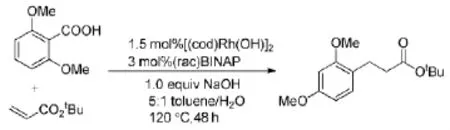
Scheme 8
Becht等[13]报道了使用催化剂三氟乙酸钯、碳酸银(1.1 equiv),以1,4-二氧六环/DMSO为溶剂,二甲氧基苯甲酸与二苯基硫醚反应得到C-S偶合产物。该反应的温度比较温和,反应的时间也有很大幅度的缩短,收率达到75%(Scheme 9)。该反应为芳环上C-S键的构建提供了一个新的方法。
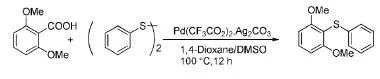
Scheme 9
但该反应不足之处是,在此反应体系中,底物适用性比较差,当甲氧基的位置发生改变(如H,I)或者被其他的基团取代(如J,K,L)时,该反应不能进行,如(Figure 4)所示。因此,也限制了该反应的应用范围。

Figure 4
Miura等[14]人研究发现,吲哚羧酸在醋酸钯和醋酸铜的催化下,与炔类化合物发生脱羧加成生成咔唑类化合物。该反应温度比较温和,为120℃。反应时间需要6 h,收率达到99%。该反应是合成多取代药物中间体咔唑的方法之一(Scheme 10)。除用于染料、医药和农药合成的传统用途外,咔唑及其衍生物被广泛用于合成光电功能材料。

Scheme 10
Lee等[15]研究发现:N-甲基吲哚-3-羧酸在有机金属钯的催化下,以二甲基苯胺为溶剂,醋酸钾为碱,与对溴苯腈发生脱羧偶联反应,得到吲哚2位的偶联产物,但收率仅为53%(Scheme 11)。

Scheme 11
Lee发现:当羧酸换成N-甲基吲哚-2-羧酸时,得到偶联的混合产物,收率达到61%,但2位偶合产物与3位偶合产物的比例为3:1(Scheme 12)。表明该反应具有一定的区域选择性。

Scheme 12
1.2分子内脱羧偶联反应
Glorius等[16]研究发现:在三氟乙酸钯和碳酸银的催化下,以DMSO和1,4-二氧六环为溶剂,2-苯氧基苯甲酸发生分子内的脱羧偶联反应。反应温度为150℃,反应时间14 h(Scheme 13)。该反应有较高的选择性,目标产物双苯并呋喃的产率高达85%。
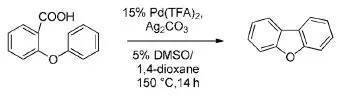
Scheme 13
反应机理:首先是碳酸银参与脱羧反应形成有机银中间体,然后经过金属交换反应和碳氢活化步骤,最后通过还原消除反应得到目标产物,同时释放出Pd0,Pd0被碳酸银氧化为PdⅡ重新参与反应,从而实现催化剂的循环,如(Figure 5)所示。
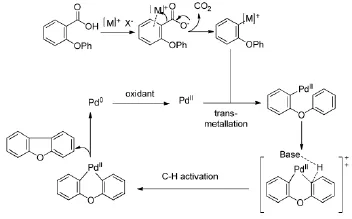
Figure 5
沈增明等[17]研究发现:2-[2-(溴苯基)甲氧基]苯甲酸在醋酸钯和三苯基膦催化下,以NMP为溶剂,碳酸钾为碱,发生分子内脱羧偶联。该反应温度较低,仅为75℃,反应时间为12 h,收率高达96%(Scheme 14)。羧酸苯环上的强吸电子基对该反应有很大影响。

Scheme 14
Forgion等[18]研究发现,药物中间体噻吩并异喹啉类化合物可以通过分子内脱羧偶联合成。底物M在氯化钯和三叔丁基膦四氟硼酸的催化下,以二甲基苯胺为溶剂,碳酸锶为碱,发生脱羧偶联反应得到化合物N,收率达到68%。化合物N又可以在有机金属钯的催化下,经过C-H活化生成化合物O。经过进一步研究发现:在以底物M合成化合物N的路线中,再加入三甲基乙酸和溴苯,通过一锅法直接合成化合物O,收率达到80%,提高了该反应的总收率(Scheme 15)。该方法是脱羧偶联反应与C-H活化反应的有机结合,极大地缩短了合成噻吩并异喹啉化合物的步骤,符合绿色化学的发展方向。

Scheme 15
2 小结
目前,脱羧偶联反应大多数用于碳碳键的构建,对于碳杂键的构建,报道的相关文献较少。羧酸及其衍生物是一类制备容易、廉价的有机合成原料。脱羧反应的主要副产物为二氧化碳,因而是一种环境友好的有机合成反应。脱羧偶联反应的这些优势引起了科学家们的关注。随着新型催化剂的深入研究与应用,脱羧偶联反应的条件越来越温和,反应收率和选择性越来越高,底物适用范围越来越广,这一方法会越来越多地应用于有机合成中。
[1](a)Torborg C,Beller M.Recent applications of palladiumcatalyzed coupling reactions in the pharmaceutical,agro chemical,and fine chemical industries[J].Advanced Synthesis&Catalysis,2009,351:3027-3043.(b)McGlacken G P,Fairlamb I J S.Palladium-catalysed cross-coupling and related processes:some interesting observations that have been exploited in synthetic chemistry[J].European Journal of Organic Chemistry,2009,24:4011-4029. (c)Nicolaou K C,Bulger P G,Sarlah D.Palladium-catalyzed cross-coupling reactions in total synthesis[J].Angewandte Chemie International Edition,2005,44:4442-4489.(d)Trost B M,Vranken D L.Asymmetric tramsition metal-catalyzed allylic alkylations[J].Chemical Re views,1996,96:395-442.
[2](a)Negishi E,Wang G,Rao H,Xu Z.Alkyne elementometalation-Pd-catalyzed cross-coupling toward synthesis of all conceivable types of acyclic alkenes in high yields,efficiently,selectively,economically,and safely:“green”way[J].Journal of Organic Chemistry,2010,75:3151-3182. (b)Terao J,Kambe N.Cross-ccoupling reaction of alkyl halides with grignard reagents catalyzed by Ni,Pd,or Cu complexes with π-carbon ligand(s)[J].Accounts of Chemical Research,2008,41:1545-1554.(c)Martin R,Buchwald S L.Palladium-catalyzed suzuki-miyaura crosscoupling reactions employing dialkylbiaryl phosphine ligands[J].Accounts of Chemical Research,2008,41:1461-1473.
[3]Lyons T W,Sanford M S.Palladium-catalyzed ligand-directed C-H dunctuinalization reactions[J].Chemical Reviews,2010,110:1147-1169.
[4]Nilsson M A.A new biaryl synthesis illustrating a connection between the ullmann biaryl synthesis and coppercatalysed decarboxylation[J].Acta Chemica Scandinavica,1966,20:423-426.
[5]Goossen L J,Deng G J,Levy L M.Synthesis of biaryls via catalytic decarboxylative coupling[J].Science,2006,313:662-664.
[6]Goossen L J,Rodriguez N,Deng G J.Biaryl synthesis via Pd-catalyzed decarboxylative coupling of aromatic carboxylates with aryl halides[J].Journal of the American Chemical Society,2007,129:4824-4833.
[7]Goossen L J,Zimmermann B,Knauber T.Palladium/copper-catalyzed decarboxylative cross-coupling of aryl chlorides with potassium carboxylates[J].Angewandte Chemie International Edition,2008,47:7103-7106.
[8]Goossen L J,Rodriguez N,Melzer B,Linger C.Enantioselective chemisorption of propylene oxide on a 2-butanol modified Pd(Ⅲ)surface:the role of hydrogen-bonding interactions[J].Journal of the American Chemical Society,2008,130:15240-15249.
[9]Goossen L J,Rodriguez N,Linder C.Decarboxylative biaryl synthesis from aromatic carboxylates and aryl triflates[J].Journal of the American Chemical Society,2008,130:15248-15249.
[10]Goossen L J,Bhadra S,Wojciech I D.Decarboxylative etherification of aromatic carboxylic acids[J].Journal of the American Chemical Society,2012,134:9938-9941.
[11]Voutchkova A,Coplin A,Leadbeater N E,et al.Palladium-catalyzed decarboxylative coupling of aromatic acids with aryl halides or unactivated arenes using microwave heating[J].Chemical Communications,2008,47:6312-6314.
[12]Ugrinov A,Pillai A F X,Sun Z M,et al.Methoxy-directed aryl-to-aryl 1,3-rhodium migration[J].Journal of the American Chemical Society,2013,135:17270-17273. [13]Becht J M,Drian C L.Formation of carbon-sulfur and carbon-selenium bonds by palladium-catalyzed decarboxylative cross-couplings of hindered 2,6-dialkoxybenzoic acids[J].Journal of Organic Chemistry,2011,76: 6327-6330.
[14]Yamashita M,Hirano K,Satoh T,et al.Synthesis of condensed heteroaromatic compounds by palladium-catalyzed oxidative coupling of heteroarene carboxylic acids with alkynes[J].Organic Letters,2009,11:2337-2340.
[15]Lee H M,Nandi D,Lee J Y.Pd(0)-catalyzed decarboxylative coupling and tandem C-H arylation/decarboxylation for the synthesis of heteroaromatic biaryls[J].Journal of Organic Chemistry,2012,77:9384-9390.
[16]Wang C Y,Piel I,Glorius F.Palladium-catalyzed intramolecular direct arylation of benzoic acids by tandem decarboxylation/C-H activation[J].Journal of the American Chemical Society,2009,131:4194-4195.
[17]Shen Z M,Ni Z J,Mo S,et al.Palladium-catalyzed intramolecular decarboxylative coupling of arene carboxylic acids/esters with aryl bromides[J].Chemistry A European Journal,2012,18:4859-4865.
[18]Forgione P,Chen F,Nicholas W Y.One-pot tandem palladium-catalyzed decarboxylative cross-coupling and C-H activation route to thienoisoquinolines[J].Advanced Syn thesis&Catalysis,2014,356:1725-1730.
Recent Development of Decarboxylative Coupling of Aromatic Carboxylic Acid
LI Yong-kai,XIE Yuan-yuan*
(College of Pharmaceutical Sciences,Zhejiang University of Technology,Hangzhou,Zhejiang 310014,China)
Carbon-carbon or carbon-heteroatom bonds can be formed by decarboxylation of carboxylic acid compounds.Decarboxylative coupling reaction ensures the regioselectivity and a major by-product is carbon dioxide.Thus,decarboxylative coupling reactions which are inexpensive,environmentally friendly meet the requirement of green chemistry.This review mainly summarized the decarboxylative coupling reactions of aromatic carboxylic acid in the past few years.
aromatic carboxylic acid;decarboxylation;coupling
1006-4184(2016)7-0005-05
2016-01-29
李永凯(1989-),男,在读硕士研究生,研究方向:药物中间体合成。
*
谢媛媛,E-mail:xyycz@zjut.edu.cn。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
建材发展导向(2021年7期)2021-07-16
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
建材发展导向(2021年24期)2021-02-12
汕头大学学报(自然科学版)(2020年4期)2020-12-14
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年5期)2017-06-13
中国塑料(2015年2期)2015-10-14
中国塑料(2015年8期)2015-10-14
股市动态分析(2015年12期)2015-09-10
