
NHO催化二氧化碳与环氧化物反应生成环碳酸酯的机理研究
2016-08-08 03:45魏明闫智娥仝文婷张祥
常州工学院学报 2016年3期
魏明,闫智娥,仝文婷,张祥
(1.南京医科大学康达学院,江苏连云港222000;2.山西师范大学化学与材料科学学院,山西临汾041001)
NHO催化二氧化碳与环氧化物反应生成环碳酸酯的机理研究
魏明1,闫智娥2,仝文婷1,张祥2
(1.南京医科大学康达学院,江苏连云港222000;2.山西师范大学化学与材料科学学院,山西临汾041001)
摘要:采用B3LYP的方法,6-311G(d,p)的基组,考虑PCM模型的溶剂化效应(DCM)以及DFT-D3模型的色散校正,对氮杂卡宾烯(NHO)催化二氧化碳和环氧化物生成环碳酸酯的反应机理进行了详细研究和讨论。该反应包括2个反应机理(M-1和M-2)。机理M-1由3个反应步骤组成,二氧化碳的加成反应是无势垒的,其中最后一步的势垒(168.74 kJ/mol)最高,是整个反应的决速步。机理M-2包括4个反应步骤,第3步具有最高势垒136.98 kJ/mol,是整个路径的最高鞍点。机理M-1和M-2在动力学上是相互竞争的,而由于机理M-2的决速步势垒比机理M-1的低,所以机理M-2是较优机理。
关键词:氮杂卡宾烯;二氧化碳;环氧化物;环碳酸酯;机理
0前言
二氧化碳的固定一直备受关注,将二氧化碳转化为有用的有机化合物不仅对化学工业有很重要的意义,同时能缓解能源危机、温室效应等问题[1]。近年来,活化二氧化碳的研究取得了较大的进展,越来越多的催化剂被发现并应用于二氧化碳的活化。作为一种二氧化碳的固定方法,二氧化碳与环氧化物反应转化为环碳酸酯,具有较好的研究前景[2]。
环碳酸酯可以被广泛用作极性非质子溶剂[3]、锂离子电池电解液[4]、机动车辆的抗爆剂以及有机合成的中间体等[5]。用于催化二氧化碳与环氧化物转化为环碳酸酯的催化剂也多种多样,如碱金属卤化物[6]、离子液体[7-9]、金属及过渡金属配合物[10]、金属氧化物、季铵盐[11]、功能高聚物[12]、金属卟啉[13]、非金属催化剂[14]等。例如:文献[15]曾报道双金属Al配合物催化二氧化碳和环氧化物的环加成反应;文献[16]报道了氨基三酚基配位的Al络合物催化二氧化碳和环氧化物合成环碳酸酯,具有较好的催化效率。
另外,非金属催化剂不仅廉价易得,而且相比金属催化剂污染小,最关键是它们具有比金属或金属配合物催化剂更高的催化活性。例如:文献[17]报道了NHC-CO2加合物和salenAlEt催化二氧化碳和环氧化物的加成,实验结果表明在相同的反应条件下,salenAlEt独自作为催化剂时不能有效催化该反应,而NHC-CO2的催化活性要比salenAlEt高很多,并且在有salenAlEt作为助催化剂时反应速率有很大的提高;文献[18]报道了用催化剂Br或NBS催化二氧化碳和环氧化物反应生成环碳酸酯,催化效率高,产率可达到92%;文献[19]用苄溴和DMF共同作为催化剂催化二氧化碳和环氧化物的反应,反应条件温和,催化效率极高,产率高达99%。显然,非金属催化剂对活化二氧化碳具有更高的活性。
近来,文献[20]报道了二氧化碳和环氧化物在氮杂卡宾烯NHO的催化下成功合成环碳酸酯的实例。虽然关于二氧化碳活化的机理已经被广泛研究,二氧化碳和环氧化物反应生成环碳酸酯的反应机理也有过很多被研究的先例,但NHO作为催化剂催化该反应的机理还未被研究。本文将进一步对NHO催化二氧化碳和环氧化物生成环碳酸酯的机理进行详细的理论研究。
1计算方法
本文使用GAUSSIAN09程序,在B3LYP理论水平上,文献[21-23]对所有参与反应的反应物、中间体以及过渡态的机构使用6-311G(d,p)基组进行了全优化。在计算过程中,选用2-甲基-环氧丙烷作为环氧化物的代表模型参与反应,节约了计算资源。用频率计算来确定所有中间体结构没有虚频,而过渡态有唯一虚频。
笔者对过渡态进行了内禀反应坐标(IRC)[24]计算,以此来验证过渡态的正确性及所连接的正、逆两方向连接着2个所期待的最小值。由于NHO催化二氧化碳和环氧化物反应生成环碳酸酯的实验研究中明确提到所使用的溶剂,因此在本文研究机理的过程中,对所有结构的优化和频率计算都是在气相中进行的。此基础上,在溶剂DCM中用对该反应中的所有结构进行了溶剂化校正(PCM模型)[25-28]。另外,将所计算的能量中增加了D3模型的经验色散校正[29]。各物质的相对能量通过下式计算所得:
GDCM=[EPCM+(Ggas-E)]× 627.509 5+Edis
(1)
ΔG=(Gsol)X-(Gsol)0
(2)
式中:GDCM表示在DCM溶液中各化合物的吉布斯自由能;EPCM表示各化合物在DCM溶液中用PCM模型计算的IEF-PCM能量;(Ggas-E)表示各化合物在气相中的吉布斯自由能和电子能的差值;Edis表示色散力作用的校正能量;ΔG表示各化合物在DCM溶液中的相对吉布斯自由能。
2结果与讨论
笔者对氮杂卡宾烯NHO催化二氧化碳和环氧化物反应生成五元环碳酸酯的机理进行了较为详细的研究。首先,用密度泛函的理论方法验证了吕小兵等提出的M-1机理,然后通过理论计算提出了新的机理M-2。其机理图的表述如图1。
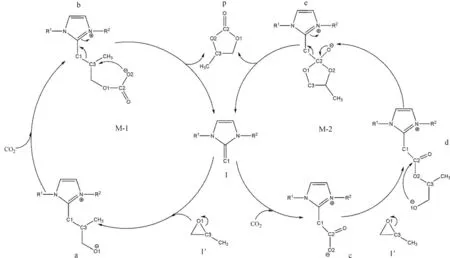
图1 NHO催化CO2和环氧化物生成五元环碳酸酯p的机理图(M-1和M-2)
反应的相对吉布斯自由能势能图如图2所示。
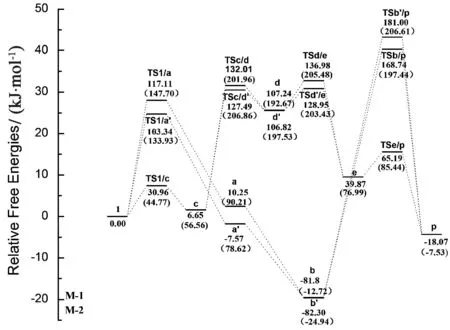
图2 在CH2Cl2(DCM)溶剂中校正后的相对吉布斯自由能势能图
考虑到反应的实际条件,选择参与反应的所有结构在DCM溶剂中校正过的能量进行讨论,在气相中的能量作为参考在图中用括号表示出来。其中,所列出的能量值是相对于反应物(NHO+CO2+2-甲基-环氧丙烷)的相对吉布斯自由能。图3分别给出了M-1,M-2所涉及的所有反应物、中间体、过渡态及产物的优化几何结构及其主要的几何参数。
2.1机理M-1
通过理论计算可以得出,正如吕小兵等提出的机理中所描述的,机理M-1的第1步是催化剂NHO和环氧化物2-甲基-环氧丙烷的开环加成反应,即具有较高电负性的催化剂氮杂卡宾烯烃1的末端碳原子C1进攻2-甲基-环氧丙烷1′的C3原子形成C1-C3键,同时C3-O1键断裂,使环氧丙烷开环从而生成开环的化合物a,这一步的过渡态是TS1/a。第2步,由于化合物a中的O1原子带有负电荷,O1原子进攻二氧化碳的显正电性的C2原子,通过形成O1-C2键,二氧化碳加成到化合物a上生成化合物b。从图3中化合物b的结构图可以看出,加成到化合物b上的CO2部分的C2-O2键长(1.24 Å)比自由二氧化碳中的C-O键的键长(1.16 Å)长0.08 Å,键角O2-C2-O变为134.9°,这说明二氧化碳已经被活化了。第3步,通过过渡态TSb/p,化合物b中的O2原子进攻C3原子,形成C3-O2键的同时C1-C3键断裂,释放出催化剂NHO,并生成最终目标产物五元环碳酸酯p。
从2-甲基-环氧丙烷的不对称性可考虑到,在机理M-1中第1步,催化剂NHO的末端碳原子C1还可以进攻2-甲基-环氧丙烷三元环上的另一个碳原子C4,C1-C4键形成的同时C4-O1键断裂,通过过渡态TS1/a′生成化合物a′。接下来,化合物a′和二氧化碳加成通过形成O1-C2键从而生成化合物b′。最后一步中通过过渡态TSb′/p,O2原子进攻C4原子形成C4-O2键,以此同时C1-C4键断裂生成五元环碳酸酯p并释放出催化剂NHO。
可以看出,催化剂NHO进攻C3或C4原子只是位阻选择性的不同,反应机理是完全相同的,生成的也是同一产物。因此,机理示意图中没有表示出TS1/a′、a′、b′、TSb′/p,而全优化的结构示意图及其主要的几何参数表示在图3中。
由图2可以看出:在DCM溶剂中,机理M-1的第1步的势垒高度是117.11 kJ/mol。中间体a相对于反应物的势能差是10.25 kJ/mol。在寻找第2步的过渡态时,始终找不到精确的过渡态结构。因此,沿O1-C2键进行柔性势能面扫描,扫描结果见图4。
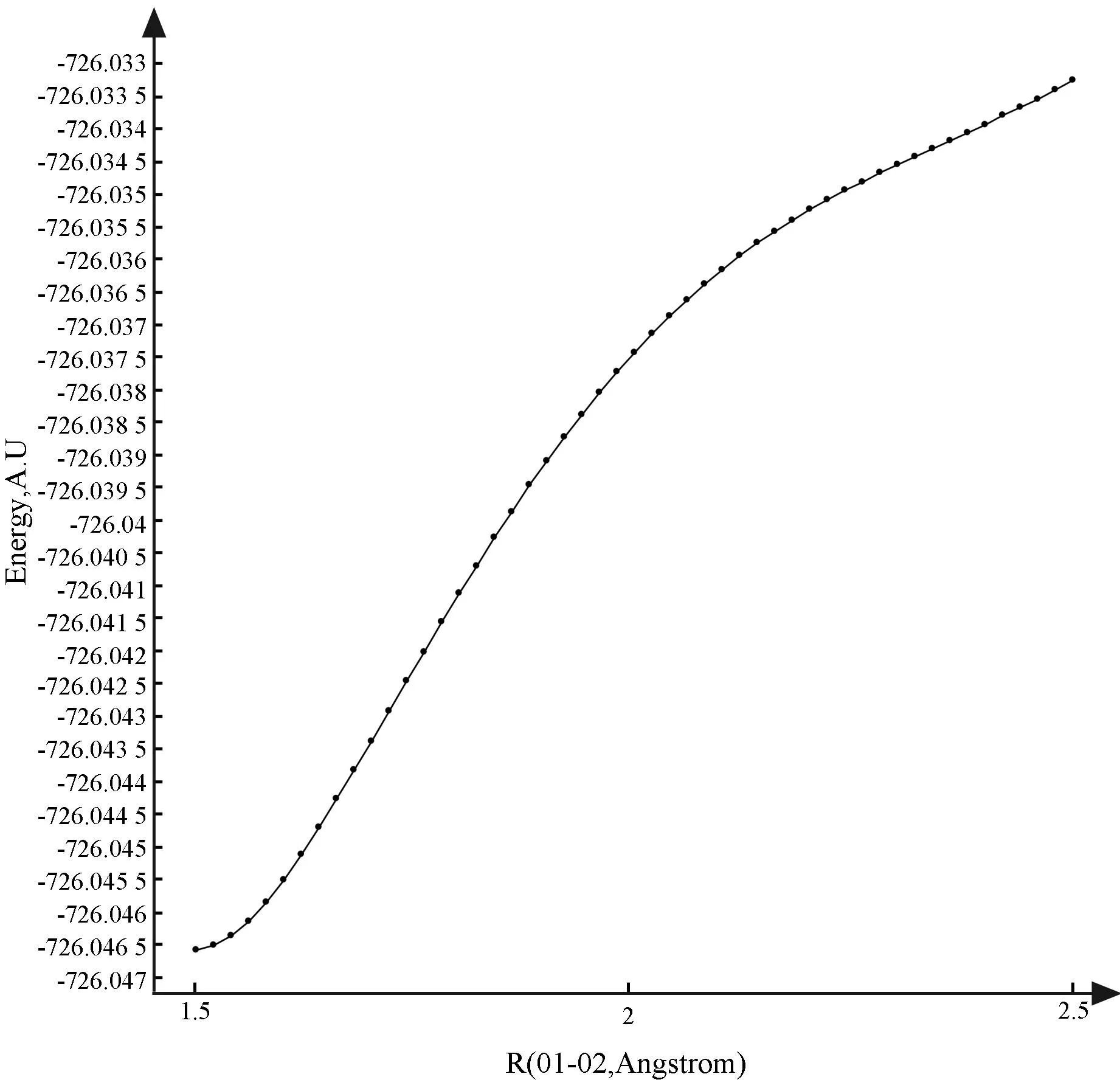
图4 沿化合物a中的O1-C2键的柔性势能面扫描(PES)
势能图显示,随着键长的增加,势能是单调递增的。由此说明,二氧化碳加成到化合物a上是一个无势垒过程,所生成中间体b的相对能量是-81.80 kJ/mol。从化合物b到最终产物p具有一个较高的反应势垒。
如图2所示,在溶剂DCM中,相对于初始反应物的能量和,其能量是168.74 kJ/mol。而这一步的活化能是250.54 kJ/mol。从整个反应路径的势能图来看,TSb/p是这条路径中的最高鞍点,说明最后一步在形成五元环的同时释放出催化剂NHO的过程需要越过的能垒是较高的。
从吕小兵等文献中给出的反应条件[20]可以看出,该反应在120 ℃下进行,在此温度下,可以越过TSb/p这个势垒。因此,机理M-1在实验温度下是一个可行的机理。从能量图可以看出,整个反应是一个放热反应,放出热量18.07 kJ/mol。
考虑位阻效应,通过理论计算总结了当NHO进攻位阻较小的C4原子时整个机理的相对吉布斯自由能。其势能图同样表示在图2中。可以看出,第1步中,TS1/a′比TS1/a的势垒高度低13.77 kJ/mol。中间体a′的相对能量是-7.57 kJ/mol,中间体a′和二氧化碳加成生成中间体b′是没有势垒的。而中间体b′的相对能量值是-82.30 kJ/mol,与b的相对能量(-81.80 kJ/mol)相近。最后一步中过渡态TSb′/p的相对能量是181.00 kJ/mol,比TSb/p的相对能量(168.74 kJ/mol)高12.26 kJ/mol。
从整个能量图可以看出,由于进攻C4原子位阻较小,反应开始时第1步的势垒高度比进攻C3原子时低,而对于反应的最后一步,势垒却比进攻C3原子时高。由于反应的决速步骤是最后一步,所以反应开始时进攻C3原子比进攻C4原子有优势。
2.2机理M-2
机理M-2的第1步是二氧化碳通过过渡态TS1/c加成到催化剂氮杂卡宾烯NHO的末端碳原子C1上形成C1-C2键,生成二氧化碳的加合物c(NHO-CO2)。在化合物c中,C1-C2和 C2-O2的键长分别是1.69 Å和1.23 Å,其中二氧化碳部分的键角O2-C2-O是136.1°,C2-O2键长比在自由二氧化碳分子中成双键时的键长伸长了0.07 Å,说明二氧化碳已经被催化剂NHO活化。
第2步是催化剂NHO与2-甲基-环氧丙烷的加成开环反应,化合物c的末端碳原子O2带有负电性,可进攻2-甲基-环氧丙烷1′的C3原子形成化合物新的O2-C3键,通过过渡态TSc/d环氧化物1′开环并加成到化合物c上,从而生成化合物d。
第3步,在化合物d中,带有负电荷的O1原子将进攻C2原子,但是由于O1原子和C2原子之间的距离较远不易成键,因此化合物d需要绕C3-C4键进行旋转使二面角O2-C3-C4-O1减小,从而缩短了O1和C2原子之间的距离进而形成O1-C2键,C2-O3的双键打开,形成1个五元环化合物e,这一步是通过过渡态TSd/e完成的。
最后一步是一个协同步骤,通过TSe/p,C2-O3双键恢复的同时C1-C2键断开,释放出催化剂NHO并生成目标产物五元环碳酸酯p。
同样考虑位阻效应,化合物c首先通过过渡态TSc/d′进攻C4原子生成化合物d′,再通过1个分子内旋转的过渡态生成五元环中间体e。从化合物e经过TSe/p到最后生成目标产物这2个结构并没有位阻效应。而反应机理与进攻C3原子时完全相同,为避免重复,并未将TSc/d′、 d′、 TSd′/e在机理图中表示出来。而将其优化的几何结构及其几何参数表示在图5中。
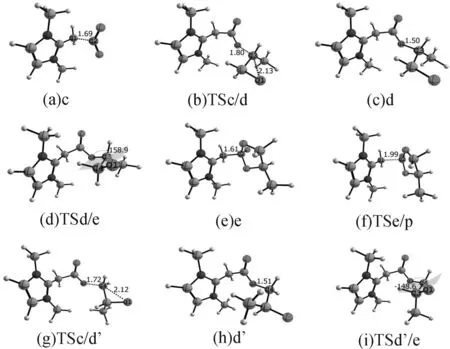
图5 机理M-2中所包含的所有中间体、过渡态及产物的结构及其重要的几何参数(键长单位:Å,键角单位:°)
从能量图2可以看出,在DCM溶剂中,反应第1步NHO与二氧化碳的加成反应需要吸收6.65 kJ/mol的热量,势垒高度是30.96 kJ/mol,这说明第1步是很容易进行的。
反应的第2步是NHO与2-甲基-环氧丙烷的开环加成反应,过渡态TSc/d的相对能量是132.01 kJ/mol,需要吸收的热量是100.58 kJ/mol,所生成中间体d的能量是107.24 kJ/mol。从中间体d到e,反应需要越过的势垒(136.98 kJ/mol)分别比第2步和最后一步高出4.98 kJ/mol和71.80 kJ/mol,这一步放出热量是67.36 kJ/mol。
最后一步中,化合物e经过TSe/p释放出催化剂和目标产物的势垒是65.19 kJ/mol,从机理M-2的整个能量图来看,第3步在DCM溶剂中的势垒最高,因此第3步是反应的决速步骤。
同样,将考虑位阻效应后总结的吉布斯自由能总结在图2中。从能量图中可以看出,TSc/d′、d′、TSd′/e的相对能量分别是127.49、106.82、128.95 kJ/mol,分别与TSc/d 、d、TSd/e的相对能量相近,说明位阻效应不是明显影响反应的势垒高度,进攻环丙烷的C3原子或C4原子是相互竞争的。
从以上讨论可以得出:对于整个反应,机理M-2是比较优越的。在吕小兵等的实验条件下(120 ℃,2 MPa),机理M-1和M-2相互竞争,而机理M-2的最高势垒(136.98 kJ/mol或128.95 kJ/mol)低于M-1的(168.74 kJ/mol或181.00 kJ/mol),因此机理M-2是比较有具竞争力的,是整个反应中的最优机理。
虽然机理M-2的最高势垒比较高,但是在实验温度120 ℃下,该势垒还是很容易可以越过。
通过比较所生成产物与初始反应物的相对能量,从能量图中可看出,该反应总体上是放热反应,所放出的热量是18.07 kJ/mol,该热量可为下一次的催化反应循环提供能量。
3结论
本章用B3LYP的方法,6-311G(d,p)的基组,结合PCM中的IEFPCM模型的溶剂化效应(DCM)以及色散校正模型DFT-D3,对二氧化碳和环氧丙烷生成环碳酸酯的反应机理进行了较为详细的研究和讨论,研究结果如下说明。
1)机理M-1由3步组成。第1步是催化剂NHO与2-甲基-环氧丙烷的开环加成反应;第2步是CO2的加成反应;第3步形成五元环碳酸酯并释放催化剂。这3步中,第1步的势垒是最低的,第2步通过柔性势能面扫描被证实是无势垒反应,最后一步中,化合物b经过TSb/p释放出催化剂和目标产物的势垒是168.74 kJ/mol(TSb′/p的相对能量是181.00 kJ/mol),是整个反应的决速步骤。
2)机理M-2包括4个反应步骤。第1步是二氧化碳与催化剂NHO的加成反应;第2步是2-甲基-环氧丙烷的开环加成反应;第3步是形成五元环的过程;第4步释放出催化剂和目标产物。第3步具有最高势垒(TSd/e和TSd′/e的相对能量分别是136.98 kJ/mol和128.95 kJ/mol),这是机理M-2在DCM溶剂中的最高鞍点,是反应的决速步骤。
3)对于整个反应有2个反应机理(M-1和M-2),这2条路径最高鞍点的相对能量比初始反应物分别高168.74、136.98 kJ/mol或181.00、128.95 kJ/mol。因此,机理M-1和M-2是相互竞争的,机理M-2是整个反应的最优机理。
[参考文献]
[1]DAI WL,LUO SL,YIN SF,et al.The direct transformation of carbon dioxide to organic carbonates over heterogeneous catalysts[J].Appl Catal ,2009,366(1): 2-12.
[2]ZHANG Y,YIN S,LUO S,et al.Cycloaddition of CO2to epoxides catalyzed by carboxyl-functionalized imidazolium-based ionic liquid grafted onto cross-linked polymer[J].Ind Eng Chem Res,2012(10): 3951-3957.
[3]WANG JQ,SUN J,CHENG WG,et al.Experimental and theoretical studies on hydrogen bond-promoted fixation of carbon dioxide and epoxides in cyclic carbonates[J].Phys Chem Chem Phys,2012(31): 11021-11026.
[4]PESCARMONA P P,TAHERIMEHR M.Challenges in the catalytic synthesis of cyclic and polymeric carbonates from epoxides and CO2[J].Catal Sci Technol,2012(11):2169-2187.
[5]YU K M K,CURCIC I,GABRIEL J,et al.Catalytic coupling of CO2with epoxide over supported and unsupported amines[J].J Phys Chem A,2010(11):3863-3872.
[6]REN Y,GUO CH,JIA JF,et al.A computational study on the chemical fixation of carbon dioxide with epoxide catalyzed by LiBr salt[J].J Phys Chem A,2011(11):2258-2267.
[7]SUN J,FUJITA SI,ARAI M.Development in the green synthesis of cyclic carbonate from carbon dioxide using ionic liquids[J].J Org Chem,2005(44):3490-3497.
[8]REN Y,MENG TT,JIA J,et al.A computational study on the chemical fixation of carbon dioxide with 2-aminobenzonitrile catalyzed by 1-butyl-3-methyl imidazolium hydroxide ionic liquids[J].Comput Thero Chem,2011(1-3):47-56.
[9]YANG ZZ,HE LN,MIAO CX,et al.Lewis basic ionic liquids-catalyzed conversion of carbon dioxide to cyclic carbonates[J].Adv Synth Catal,2010(13):2233-2240.
[10]SIBAOUIH A,RYAN P,LESKELM,et al.Facile synthesis of cyclic carbonates from CO2and epoxides with cobalt(II)/onium salt based catalysts[J].Appl Catal A,2009(2):194-198.
[11]CALO V,NACCI A,MONOPOLI A,et al.Cyclic carbonate formation from carbon dioxide and oxiranes in tetrabutylammonium Halides as Solvents and Catalysts[J].Org Lett,2002(48):2561-2563.
[12]XIE Y,ZHANG Z,JIANG T,et al.CO2cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross-linked polymer matrix[J].Angew Chem Int Edit,2007(38):7255-7261.
[13]BAI D,WANG X,SONG Y,et al.Bifunctional metalloporphyrins-catalyzed coupling reaction of epoxides and CO2to cyclic carbonates[J].Chin J Catal,2010(2):176-180.
[14]AJITHA M J,SURESH C H.NHC catalyzed CO2fixation with epoxides: Probable mechanisms reveal ter molecular pathway[J].Tetrahedron Lett,2011(41):5403-5406.
[15]TIAN D,LIU B,GAN Q,et al.Formation of cyclic carbonates from carbon dioxide and epoxides coupling reactions erfficiently catalyzed by robust,recyclable one-component aluminum-salen complexes[J].Acs Catal,2012(2):2029-2035.
[16]WHITEOAK C J,KIELLAND N,LASERNA V,et al.A powerful aluminum catalyst for the synthesis of highly functional organic carbonates[J].J Am Chem Soc,2013,135(4):1228-1231.
[17]ZHO H,ZHANG W Z,LIU C,et al.CO2adducts of N-heterocyclic carbenes:Thermal stability and catalytic activity toward the coupling of CO2with epoxides[J].J Org Chem,2008(20):8039-8044.
[18]KOZAK J A,WU J,SU X,et al.Bromine-catalyzed conversion of CO2and epoxides to cyclic carbonates under continuous flow conditions[J].J Am Chem So,2013,135(49):18497-18501.
[19]WANG L,LIN L,ZHANG G,et al.Synthesis of cyclic carbonates from CO2and epoxides catalyzed by low loadings of benzyl bromide/DMF at ambient pressure[J].Chem Commun.,2014(94):14813-14818.
[20]WANG YB,SUN DS,ZHOU H,et al.CO2,COS and CS2adducts of N-heterocyclic olefins and their application as organocatalysts for carbon dioxide fixatio[J].Green Chem,2015(7):4009-4015.
[21]STEPHENS P J,DEVLIN J,CHABALOWSKIC F C,et al.Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields[J].J Phys Chem,1994(45):11623-11627.
[22]LEE C,YANG W,PARR R G.Development of the colic-salvetti correlation-energy formula into a functional of the electron density[J].Phys Rev B,1988(2):785-789.
[23]BECKE A D.Density-functional thermochemistry.III:The role of exact exchange[J].J Chem Phys,1993(7):5648-5653.
[24]GONZALEZ C,SCHLEGEL H B.Reaction path following in mass-weighted internal coordinates[J].J Phys Chem,1990(14):5523-5527.
[25]MIERTUS S,SCROCCO E,TOMASI J.Electrostatic interaction of a solute with a continuum:A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects[J].Chem Phys,1981(1):117.
[26]PASCUAL-AHUIR J L,SILLA E,TOMASI J,et al.Electrostatic interaction of a solute with a continuum:Improved description of the cavity and of the surface cavity bound charge distribution[J].J Comput Chem,1987(8):778.
[27]TOMASI J,PERSICO M.Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent[J].Chem Rev,1994(7):2027.
[28]FLORIS F,TOMASI J.Evaluation of the dispersion contribution to the solvation energy:A simple computational model in the continuum approximation[J].J Comput Chem,1989(5):616.
[29]GRIMME S,ANTONY J,EHRLICH S,et al.A consistent and accurate ab initio parametrization of density functional dispersion correction(DFT-D)for the 94 elements H-Pu[J].J Chem Phys,2010(15):154104.
责任编辑:陈亮
doi:10.3969/j.issn.1671-0436.2016.03.009
收稿日期:2016- 04-19
作者简介:魏明(1988—),男,硕士,助教。
中图分类号:O641.12+1
文献标志码:A
文章编号:1671- 0436(2016)03- 0038- 07
Mechanism for NHO-Catalyzed Synthesis of Cyclocarbonates from CO2and Exopides: A DFT Study
WEI Ming1,YAN Zhie2,TONG Wenting1,ZHANG Xiang2
(1.Kangda College of Nanjing Medical University,Lianyungang 222000;2.School of Chemistry and Material Science,Shanxi Normal University,Linfen 041001)
Abstract:The mechanisms for N-heterocyclic Olefin-catalyzed formation of cyclic carbonate from CO2 and epoxide were studied by means of comprehensive density functional theory calculations at the the B3LYP/6-311G(d,p) level.For inclusion of salvation effect,single point energy calculations were performed using the integral equation formalism polarizable continuum model (IEF-PCM) with radius and the static relationship taken from SMD model.In addition,the empirical dispersion correction recommended by Grimme was added to the B3LYP energies.The formation of cyclic carbonate from CO2 and epoxide contains two mechanisms (M-1 and M-2).M-1 is composed of three steps,the step of CO2 addition in M-1 is barrierless,and the final step with the highest barrier (168.74 kJ/mol) is the rate determining step of the reaction.M-2 includes four reaction steps,the third step with a barrier of 136.98 kJ/mol,is the highest saddle point of the reaction path.M-1 and M-2 may play as kinetically competing mechanisms,M-2 is the predominant one with the lower barrier for the rate controlling step.
Key words:N-heterocyclic Olefin;CO2;epoxide;cyclic carbonate;mechanism
猜你喜欢
哈哈画报(2022年8期)2022-11-23
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
建材发展导向(2021年14期)2021-08-23
小学科学(学生版)(2021年5期)2021-07-22
学生天地(2020年18期)2020-08-25
中国煤层气(2019年2期)2019-08-27
环境与可持续发展(2017年2期)2017-04-06
汽车零部件(2014年8期)2014-12-28
火炸药学报(2014年1期)2014-03-20
