
α珠蛋白基因新突变复合α地中海贫血家系的基因分析及产前诊断
2016-08-05 06:03:08王继成杜丽秦丹卿
新医学 2016年7期
王继成 杜丽 秦丹卿
α珠蛋白基因新突变复合α地中海贫血家系的基因分析及产前诊断
王继成杜丽秦丹卿
511442 广州,广东省妇幼保健院医学遗传中心 妇幼代谢与遗传病重点实验室(王继成)
【摘要】目的对一个α珠蛋白基因新突变复合α地中海贫血的家系进行基因分析和产前诊断。方法采集家系成员的外周血标本进行血红蛋白毛细管电泳和红细胞参数的分析,应用gap-PCR、PCR-RDB和DNA测序方法对外周血和绒毛标本DNA进行α珠蛋白基因的鉴定。结果检测到的α珠蛋白基因基因型为母亲--/αCSα突变,父亲--/αα复合HbA2:c.52G>T(p.Val17Phe)突变,绒毛标本提示胎儿的基因型为αCSα/αα复合HbA2:c.52G>T突变。 结论HbA2:c.52G>T是一种至今国内外未曾报道的新突变,而由其导致的异常血红蛋白可加重地中海贫血的临床症状。
【关键词】α-地中海贫血;点突变;α珠蛋白基因
血红蛋白病是全世界最常见的遗传性血液病,可分为地中海贫血(地贫)和异常血红蛋白。前者是由于珠蛋白基因缺陷导致珠蛋白肽链的合成减少,而后者表现为珠蛋白的结构异常。因为多数已发现的异常血红蛋白没有明显的临床症状,当常规地贫基因检测出阳性结果时,由点突变引起的血红蛋白病常被忽视[1]。HbVar数据库(http://globin.cse.psu.edu)显示目前全世界已发现超过700多种α珠蛋白基因的变异体,多数发生在α2珠蛋白基因上[2-3]。本研究发现一种新的α珠蛋白基因突变,并对其进行了临床分析和产前诊断,现报告如下。
对象与方法
一、 研究对象
家系中的孕妇,27岁,停经12周,血常规表现为小细胞低色素中度贫血,曾行脾脏介入栓塞术。外院常规地贫基因检测显示为--/αCSα中间型地贫。孕妇的丈夫,27岁,血常规提示为小细胞低色素轻度贫血。本院常规地贫基因检测显示为--/αα轻型地贫,血红蛋白电泳显示HbA2 0.8%、HbH 8.2%。包括血常规在内的血液学结果与常规基因检测结果不符,拟诊断其丈夫为中间型α地贫。
二、 方法
使用迈瑞2000血液分析仪对外周血进行红细胞参数分析。应用快速电泳分析系统(法国sebia capillary2)对外周血进行血红蛋白分析。采用亚能生物技术有限公司提供的试剂盒对外周血和绒毛标本进行三种最常见的缺失型α地贫(--SEA/、-α4.2、-α3.7)和三种最常见的点突变型α地贫(HbCS、HbWS、HbQS)以及17种β地贫的基因检测。设计引物分别扩增α1和α2珠蛋白基因,PCR产物送上海英潍捷基生物有限公司进行双向测序。引物序列如下:HbA1 5’-TGGAGGGTGGAGACGTCCTG-3’和5’-TCCATCCCCTCCTCCCGCCCCTGCCTTTTC-3’,HbA2 5’-GATGGGCGGGAGTGGAGT-3’和5’-GGACAGGGGATGGTTCAGC-3’,产物大小分别为1 181 bp和1 241 bp。
结果
一、 血液学结果
家系成员的血液学分析结果见表1。
二、 分子诊断结果
常规地贫基因检测和α珠蛋白基因测序的结果为检测到母亲的α珠蛋白基因基因型为--/αCSα突变,父亲为--/αα复合HbA2:c.52G>T(p.Val17Phe)突变,绒毛标本提示胎儿的基因型为αCSα/αα复合HbA2:c.52G>T突变。父亲与胎儿α2珠蛋白突变的测序图见图1A、B。
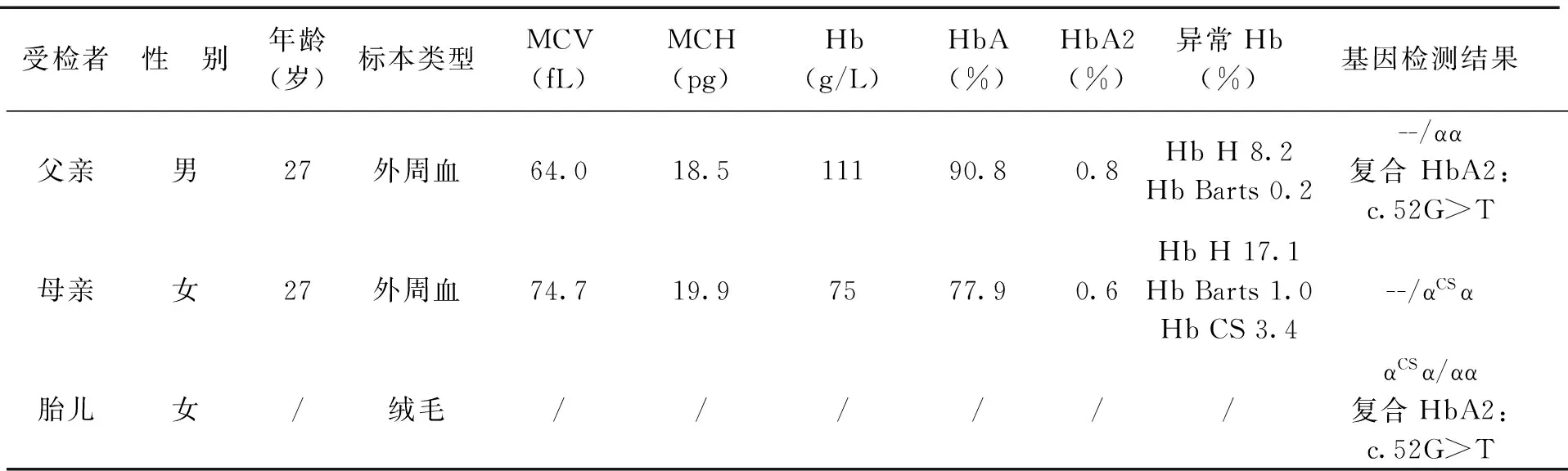
表1 家系成员的血液学参数和基因结果

图1 父亲与胎儿α2珠蛋白突变的测序图
A:父亲的HbA2:c.52G>T纯合突变测序图;B:胎儿的HbA2:c.52G>T杂合突变测序图
讨论
我国的广东、广西等华南地区是异常血红蛋白发生率较高的地区,北方地区的发生率明显低于南方。其发病率及突变类型在不同地区、不同种族的分布具有差异性[4-6]。能引起明显珠蛋白功能改变的异常血红蛋白包括地中海贫血样变异、溶血性异常血红蛋白和氧亲和力异常性异常血红蛋白病。这些异常血红蛋白可以在单独存在或复合地中海贫血时,表现为中间型或重型地中海贫血[7]。异常血红蛋白的传统鉴定传统方法采用的是珠蛋白肽链化学结构分析的方法,目前常用反相高效液相色谱(RP-HPLC)分析血红蛋白[8-9]。本研究中采用的sebia capillary 2方法进行血红蛋白分析并未发现特异性的异常血红蛋白。
据我们所知,HbA2: c.52G>T(p.Val17Phe)突变是一种至今国内外未曾报道的新突变。这种突变使α2珠蛋白基因第52位密码子由GTC变成TTC,导致缬氨酸替换为苯丙氨酸。蛋白质的一级结构决定其空间结构,而空间结构与蛋白的功能和稳定性相关。查询HbVar数据库发现,已报道的HbA2:c.53T>A(p. Val17Asp)突变与本研究报道的突变位于同一编码子,均引起编码氨基酸的改变。53T>A在杂合突变的情况下,显示正常的临床表现,这与52G>T的突变引起的珠蛋白的功能或稳定性的改变可能是不同的。因为本研究中的父亲的血液学表现明显与单纯的东南亚缺失型α地贫不同,表现为中间型α地贫。当然本突变是否会引起珠蛋白功能和稳定性的改变,需要进一步在功能学上进行鉴定。
非缺失型α地中海贫血患者的血液学表现不同于缺失型患者,贫血更为严重[10-11]。依据胎儿的基因型,我们推测胎儿是轻度至中度的地贫。因为CS突变和新发现的突变均累及功能较强的α2基因,CS产生的异常肽链对红细胞也有较强的破坏作用而引起慢性溶血性贫血[12]。从优生遗传的角度来看,此基因型的胎儿是这对夫妇可以拥有的临床表型最轻的α地贫后代,所以我们建议谨慎考虑和选择胎儿的去留。当然在遗传咨询中,对于此类基因型需加以解释说明其表型的多样性。
总之,HbA2:c.52G>T是一种新发现的突变,这丰富了血红蛋白变异体的数据库。对这种突变的详细的基因分析和临床症状的描述有赖于我们今后进行进一步的血红蛋白功能学的研究。
参考文献
[1]曾溢滔.人类血红蛋白.北京:科学出版社,2002:150-175.
[2]Hardison RC, Chui DH, Giardine B, Riemer C, Patrinos GP, Anagnou N, Miller W, Wajcman H. HbVar: a relational database of human hemoglobin variants and thalassemia mutations of the globin gene server. Hum Mutat,2002,19(3):225-233.
[3]Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, Bartsakoulia M, Riemer C, Miller W, Tzimas G, Wajcman H, Hardison RC, Patrinos GP. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res,2014,42(Database issue):D1063-D1069.
[4]秦良谊.我国异常血红蛋白发生率、分布及遗传多态性.医学研究通讯,2003,32(12):12-14.
[5]Lou JW, Wang T, Liu YH, He Y, Zhong BM, Liu JX, Zhao Y, Ye WL, Li DZ. Prevalence and molecular characterization of structural hemoglobin variants in the Dongguan region of Guangdong province, southern China. Hemoglobin,2014,38(4):282-286.
[6]Lin M, Wang Q, Zheng L, Huang Y, Lin F, Lin CP, Yang LY. Prevalence and molecular characterization of abnormal hemoglobin in eastern Guangdong of southern China. Clin Genet,2012,81(2):165-171.
[7]徐湘民.地中海贫血预防控制操作指南.北京:人民军医出版社,2011:11-13.
[8]Wan JH, Tian PL, Luo WH, Wu BY, Xiong F, Zhou WJ, Wei XC, Xu XM. Rapid determination of human globin chains using reversed-phase high-performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci,2012,901:53-58.
[9]Levi JE, da Costa Lira SM, de Carvalho Polite MB, d'Almeida Pereira RA, Mota M1, Bianco C, Kutner JM. Simultaneous blood donor screening for abnormal hemoglobin levels and glycated hemoglobin (HbA1c) by high-performance liquid chromatography. Transfusion,2015,55(9):2291-2292.
[10]陈素琴,蒋玮莹,陈路明,田秋红,曾瑞萍.中国南方地区血红蛋白 H 病的基因型与表型的相关性研究.新医学,2015, 46(5):294-298.
[11]Chao YH, Wu KH, Wu HP, Liu SC, Peng CT, Lee MS. Clinical features and molecular analysis of HbH disease in Taiwan. Biomed Res Int,2014,2014:271070.
[12]Schrier SL, Bunyaratvej A, Khuhapinant A, Fucharoen S, Aljurf M, Snyder LM, Keifer CR, Ma L, Mohandas N. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood,1997,89(5):1762-1769.
(本文编辑:杨江瑜)
DOI:10.3969/j.issn.0253-9802.2016.07.015
通讯作者,秦丹卿,E-mail:qindanqing1985@163.com
(收稿日期:2015-11-06)
Genetic analysis and prenatal diagnosis of a family with novel α-globin gene mutation complicated with α-thalassemia
WangJicheng,DuLi,QinDanqing.
MedicalGeneticsCenterofGuangdongWomenandChildrenHospital,MaternalandChildrenMetabolic-GeneticKeyLaboratoryofGuangdong,Guangzhou511442,ChinaCorrespondingauthor,QinDanqing,E-mail:qindanqing1985@163.com
【Abstract】ObjectiveTo perform genetic analysis and prenatal diagnosis of a family with novel α-globin gene mutation complicated with α-thalassemia. MethodsPeripheral blood sample was collected from the family members for hemoglobin electrophoresis and red blood cell (RBC) analysis. Gap-PCR, PCR-RDB and DNA sequencing were utilized to detectα-globin gene mutation in the peripheral blood and villi samples. ResultsThe genotype of α-globin gene of the father was --/αα complicated with HbA2:c.52G>T (p.Val17Phe), and --/αCSα for the mother. The villi specimen detected that the genotype of fetus was αCSα/αα complicated with HbA2:c.52G>T mutation. ConclusionHbA2:c.52G>T mutation has not been reported. Abnormal hemoglobin induced by HbA2:c.52G>T mutation can aggravate the clinical symptoms of thalassemia.
【Key words】α-thalassemia;Point mutation; α-globin gene
猜你喜欢
中国毕业后医学教育(2022年1期)2022-08-19 02:51:34
科学大众(2021年6期)2022-01-01 00:45:52
检验医学与临床(2021年14期)2021-07-29 07:40:24
基层中医药(2021年4期)2021-07-22 07:15:26
基层中医药(2021年4期)2021-07-22 07:15:18
家庭医学(下半月)(2020年6期)2020-08-24 07:46:14
检验医学与临床(2015年14期)2015-03-16 01:46:53
现代检验医学杂志(2015年1期)2015-02-06 01:59:18
实验动物与比较医学(2014年3期)2014-02-28 14:52:55
卫生职业教育(2014年8期)2014-02-16 08:00:16
