
HPLC法测定硫酸氢氯吡格雷有关物质
2016-08-02 19:10吴相永
卷宗 2016年6期
关键词:高效液相色谱法
吴相永
摘 要:建立高效液相色谱法测定硫酸氢氯吡格雷有关物质的方法。根据文献资料[4,5],采用高效液相色谱法,以手性色谱柱,建立了硫酸氢氯吡格雷的有关物质测定方法,方法简便,准确。采用ULTRONES-OVM(Analytical)手性柱(150mm×4.6mm,5μm),色谱填料为L57。以0.01mol/L磷酸二氢钾溶液-乙腈(75∶25)为流动相,检测波长为220nm。硫酸氢氯吡格雷和杂质B1之间的分离度大于2.5,符合要求。本方法具有简便、快捷、准确、灵敏和专属性强的特点,符合本品有关物质的测定要求。
关键词:高效液相色谱法;硫酸氢氯吡格雷;有关物质测定
1 引言
硫酸氢氯吡格雷(Clopidogrelhydrogensulfate)是一种血小板聚集抑制剂,临床上用于预防和治疗因血小板高聚集状态引起的心、脑及其他动脉的循环障碍疾病[1-3]。由法国Sanofi-Aventis原研,1998年美国上市,2001年在我国批准上市,商品名波立维( Plavix)。根据文献资料[4,5],采用高效液相色谱法,以手性色谱柱,建立了硫酸氢氯吡格雷的有关物质测定方法。
2 实验部分
2.1 仪器与试药
仪器 安捷伦1100高效液相色谱仪带二极管阵列检测器
ULTRONES-OVM手性色谱柱( 15cm×4.6mm,5μm)
梅特勒-托利多MX205分析天平
试药 硫酸氢氯吡格雷对照品( 中国药品生物制品检定所),批号:100819-200601
杂质A:USP对照品(批号:G0H250),用于杂质定量分析。
杂质B:USP对照品(批号:G1H047),用于杂质定量分析。
杂质C:USP对照品(批号:F2H215),用于杂质定量分析。
2.2 方法的建立
2.2.1检测波长的选择
将本品、杂质A、杂质B和杂质C制成适当溶液后进行紫外扫描,结果本品和已知杂质的紫外吸收行为类似,均在270nm、277nm和低波长附近处有最大吸收。由于多数物质在低波长处均有吸收,且本品与已知杂质的低波长区吸收强度远高于270nm,因此为更多的检出未知杂质,同时参考USP31版中硫酸氢氯吡格雷测定的检测波长220nm,选择低波长220nm作为有关物质的测定波长。
2.2.2色谱条件的确定
参考国家标准(WS1-(X-471)-2003Z)[4]和USP31版药典[5]的有关物质检测方法,并进行了对比研究,最终选择了本品的有关物质检测条件:采用ES-OVM柱;以0.01mol/L磷酸二氢钾溶液-乙腈(75:25)为流动相;检测波长:220nm;进样量:10μl;流速:1.0ml/min。
2.2.3系统适用性试验
按上述色谱条件进行试验,取杂质对照品溶液进样,记录色谱图,出峰顺序分别为杂质A、杂质B第一个异构体(杂质B1)、硫酸氢氯吡格雷、杂质B第二个异构体(杂质B2)和杂质C;取系统适用性试验用溶液进样,杂质B第一个异构体与硫酸氢氯吡格雷分离度不小于2.5,实际测得结果为3.3。
2.3 方法学验证与结果
2.3.1专属性试验
分别量取溶剂空白、未破坏样品、酸破坏、碱破坏、热破坏、光破坏溶液各10μl,分别注入液相色谱仪,记录色谱图。结果本品在酸、碱、热及强光照射条件下均有不同程度的破坏,新增大的降解峰主要为杂质A和杂质C,并且与硫酸氢氯吡格雷峰分离良好,可知本方法专属性高,适合对本品有关物质的检测。
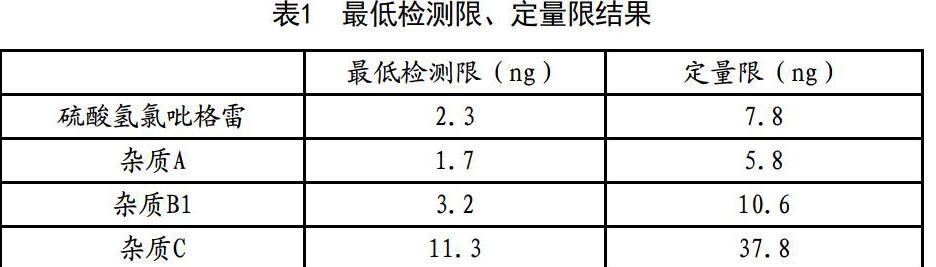
2.3.2最低检测限、定量限
取硫酸氢氯吡格雷对照品、杂质A、杂质B和杂质C贮备液各适量,分别加流动相稀释至峰响应值约为基线噪音水平3倍、10倍的浓度,精密量取各供试液10μl,注入液相色谱仪,记录色谱图,计算最低检测限和定量限。结果见表1。
2.3.3进样精密度试验
取杂质对照品溶液,连续进样6次,考察杂质A、杂质B1、硫酸氢氯吡格雷和杂质C的峰面积变化情况,结果杂质A、杂质B1、硫酸氢氯吡格雷和杂质C峰面积的RSD分别为3.0%、3.3%、1.8%、2.8%,RSD均小于10,说明本方法进样精密度良好,符合杂质定量测定的要求。
2.3.4重复性试验
取本品约100mg,精密称定,置200ml量瓶中,用甲醇5.0ml溶解后加流动相稀释成至刻度,摇匀,作为供试品溶液。分别配制6份供试品溶液,各精密量取10μl进样分析,以外标法计算已知杂质的量,考察测定结果的相对标准偏差,结果雜质A、杂质B1、杂质C峰面积的RSD分别为3.9%、5.4%、1.8%,均小于10%,说明本法重复性良好。
2.3.5线性试验
分别精密量取硫酸氢氯吡格雷对照品、杂质A、杂质B和杂质C贮备液适量,加流动相逐级稀释,得硫酸氢氯吡格雷、杂质A、杂质B1和杂质C的线性试验溶液。精密量取各溶液10?l分别进样,记录色谱图。
以峰面积A为纵坐标、浓度c为横坐标做线性回归,得杂质A、杂质C、硫酸氢氯吡格雷、杂质B1的曲线分别为:A = 21.365c -1.4165,r=0.9981,A = 15.156c-0.2251,r=0.9979,A = 17.724c + 3.8769,r=0.9995,A = 19.711c -0.1161,r=0.9995。试验结果表明杂质A在浓度0.41~4.88μg/ml,杂质B1在浓度0.6~11.46μg/ml,杂质C在浓度3.43~12.21μg/ml,硫酸氢氯吡格雷在浓度0.36~3.98μg/ml范围内,浓度与峰面积呈良好的线性关系。
2.3.6回收率试验
采用加样回收的方法对杂质A、B和C进行了回收率试验研究。
杂质A对照品母液的配制:取杂质A适量,用甲醇溶解并稀释至每1ml含杂质A约40μg的溶液,即得。
杂质B对照品母液的配制:取杂质B适量,用甲醇溶解并稀释至每1ml含杂质B约120μg的溶液,即得。
杂质C对照品母液的配制:取杂质C适量,用甲醇溶解并稀释至每1ml含杂质C约200μg的溶液,即得。
供试品溶液的配制:取本品约100mg,置200ml量瓶中,加甲醇5.0ml溶解后加流动相稀释至刻度,摇匀,作为样品贮备母液;取5ml,置10ml量瓶中,分别加杂质A对照品母液、杂质B对照品母液和杂质C对照品母液各0.8ml、1.0ml和1.2ml,得含有80%、100%和120%杂质的供试品溶液,每份各平行配制三次。
精密量取上述供试品溶液各10μl,注入液相色谱仪,记录色谱图,以外标法计算加入杂质的含量,计算回收率及相对标准偏差,结果杂质A、杂质B1、杂质C回收率分别为106.7%、106.1%、101.6%,RSD分别为4.4%、5.8%、2.7%。由上述试验结果可知,杂质A、杂质B1和杂质C的回收率良好,均在80%~120%之间,且RSD%均小于10,满足已知杂质定量测定的要求
2.3.7耐用性试验
在确定的有关物质测定方法的基础上,分别考察了流动相比例变化±5%、柱温变化±5℃和流速變化±0.2 ml/min对有关物质测定结果的影响。试验结果表面,当温度、流速和流动相比例发生略微改变时,有关物质的测定结果基本不受影响。
2.3.8有关物质测定的溶液稳定性
取本品约100mg置200ml量瓶中,加甲醇5.0ml溶解后加流动相稀释成至刻度,摇匀,室温下放置,分别于0、2、4、8小时进样测定,记录色谱图。结果表明本品的有关物质溶液在8小时内基本稳定,满足分析测定的需要。
3 讨论
3.1 本文采用手性色谱柱,能使主成分及各杂质有效分离,结果准确可靠,满足测定要求;
3.2本法测定硫酸氢氯吡格雷有关物质, 专属性强, 结果准确、 灵敏、 快速。为硫酸氢氯吡格雷的质量控制提供了值得借鉴的方法。
参考文献
[1]袁静.氯毗格雷的药理学、临床疗效和耐受性[J].中国临床药学杂志,1999,8(4):69—70.
[2]Mitakos A,PandenL.A Validated LC Method for theDetermination of Clopidogrel in Pharmaceutical Preparations[J].J.Pharm Biomed Anal., 2002, 28(3): 431—438.
[3]孙立平,张林潮.氯吡格雷在冠心病中的应用[J].医学综述, 2006, 12(19): 1190— 1192.
[4]国家药典委员会.新药转正标准硫酸氢氯吡格雷[S].WS1-(X-471)-2003Z,北京:化学工业出版社,2003.
[5]美国药典委员会.美国药典[M/CD].第32版,美国:美国光盘发行社,2008.
猜你喜欢
考试周刊(2016年103期)2017-01-23
中国民族民间医药·下半月(2016年11期)2017-01-19
中国当代医药(2016年30期)2017-01-07
中国当代医药(2016年29期)2017-01-03
中国民族民间医药·上半月(2016年11期)2016-12-26
