
阳极氧化对镁合金在仿生溶液中降解性能影响的研究*
2016-07-16 07:59:03张春艳李盼妮杨明波徐超玲
功能材料 2016年6期
关键词:镁合金
张春艳,李盼妮,杨明波,徐超玲
(重庆理工大学 材料科学与工程学院,重庆 400054)
阳极氧化对镁合金在仿生溶液中降解性能影响的研究*
张春艳,李盼妮,杨明波,徐超玲
(重庆理工大学 材料科学与工程学院,重庆 400054)
摘要:利用电化学测试技术和浸泡试验研究了几种电压下的阳极氧化工艺对镁合金在Hank’s仿生溶液中降解性能的影响, 同时对Hank’s溶液浸泡前后的氧化膜的形貌和成分进行了比较分析, 以探讨氧化膜在仿生溶液中的降解行为。研究表明,随阳极氧化电压增大,阳极氧化膜表面的孔隙增大,电压升至110 V时,试样表面烧蚀,阳极氧化膜疏松,平整度较差。阳极氧化提高了镁合金的耐蚀性,100 V下的阳极氧化试样具有最大的涂层电阻,90 V下阳极氧化试样具有最大电荷转移电阻和极化电阻。阳极氧化处理后的试样在Hank’s仿生溶液中可延缓镁合金降解1周左右,随后析氢速率有所增高。其中110 V电压下生成的阳极氧化试样自腐蚀电位降低,浸泡1周后的析氢量明显高于其它试样。浸泡过程中,随着阳极氧化层的溶解,Hank’s溶液中的磷酸根和钙离子沉积在氧化层表面形成磷酸钙盐,其产物进入氧化膜的小孔内,形成无孔表面。
关键词:镁合金;阳极氧化;降解性能;生物材料
0引言
镁及镁合金具有比重轻、比强度高以及优良的生物相容性、力学相容性和生物降解性等优点,近年来,镁基生物医用材料的研究和开发受到广泛关注。但镁的标准电极电位(-2.37 V)很低,表面氧化层存在缺陷,耐蚀性较差[1-2]。植入件腐蚀速率过快不仅不能满足骨骼生长的力学和形态学性能的要求[3],还会导致皮下析氢速率加快,造成不适和植入失败[4]。此外,腐蚀产物也会造成局部pH值过高,诱发炎症甚至细胞溶血等临床症状[5-6]。这些问题在很大程度上限制了镁合金在生物材料领域的应用。
表面处理是改善镁合金耐蚀性和生物相容性的有效途径之一。阳极氧化或微弧氧化(MAO或PEO)是一种通过施加阳极电流在金属和合金上形成稳定的氧化膜的工艺。阳极氧化膜属于陶瓷涂层,其优点是与基体具有良好的结合力,具有多孔的特点,这种多孔膜层有助于组织生长和制备载药涂层[7]。研究表明,MAO膜层可显著提高纯镁、镁合金ZK60以及镁合金AZ91D在Hank’s仿生溶液中的耐蚀性,延缓镁基体的生物降解[8-10]。郭磊等研究认为MgO膜不仅可有效地延缓AZ31B镁合金降解产物的释放速度,而且能显著降低合金的致突变反应和溶血反应,对成骨细胞的增殖和成骨活性无毒性作用[11]。Yang等在ZK60镁合金表面制备了含Mg2SiO4的微弧氧化层,该氧化层不仅可改善镁合金的耐蚀性和降解性能,而且有利于骨髓干细胞的分化,显著降低镁合金的溶血率,改善了镁合金的生物相容性和生物活性[12]。
镁合金生物材料在人体中的应用难题主要还是集中在腐蚀机理的研究和腐蚀速率的控制上。阳极氧化膜的厚度、表面状态都会影响到其降解性能,本文通过电化学测试技术和浸泡析氢试验研究了不同电压下阳极氧化对镁合金降解性能的影响,有助于理解阳极氧化膜状态和控制镁合金腐蚀速率之间的关系。
1实验
1.1试样制备
将挤压镁合金板材AZ31切割成30 mm×40 mm×1.5 mm的试样若干,依次使用200,400,600,800和1 000#金相水砂纸对镁合金试片逐级打磨光滑,蒸馏水洗净。试样先在50~60 ℃下碱洗5 min,蒸馏水冲洗后,然后在室温下酸洗1 min,最后用蒸馏水冲洗干净,干燥待用。碱洗液组成:Na3PO450 g/L;Na2SiO325 g/L;Na2CO350 g/L。 酸洗液组成:CH3COOH 195 mL/L;NaNO340 g/L。
1.2阳极氧化处理
镁合金试样作为阳极与电源的正极相连,不锈钢板作为阴极与电源的负极相连,采用国产WYK-30010直流稳压稳流电源,分别在80,90,100和110 V下制备阳极氧化膜。阳极氧化溶液为:60 g/L KOH,70 g/L Na2SiO3,60 g/L Na2B4O7, 30 g/L Na2CO3。不锈钢板与镁合金试样的面积比为2∶1,在阳极氧化过程中,磁力搅拌器不断搅拌电解液直至阳极氧化结束,使得形成均匀的阳极氧化膜层。
1.3析氢实验
将镁合金AZ31和阳极氧化后的试样浸入500 mL的Hank’s溶液中进行腐蚀析氢实验,试样总的表面积约为10 cm2。实验温度为室温(32~34 ℃)。每天记录两次滴定管液面的变化,计算析出氢气的体积。15 d后取出试样,绘制析氢体积-时间曲线,并观察试样表面的形貌。
1.4电化学测试
电化学性能测试采用CHI604C电化学工作站,采用三极体系进行测试,腐蚀介质Hank’s溶液(8.0 g/L NaCl,0.4 g/L KCl,0.14 g/L CaCl2,0.35 g/L NaHCO3,1.0 g/L C6H12O6,0.1 g/L MgCl2·6H2O,0.06 g/L MgSO4·7H2O,0.06 g/L KH2PO4,0.06 g/L NaHPO4·12H2O),测试过程在室温下进行。测量前,试样先于Hank’s溶液中静置10 min使其达到稳态。然后进行极化曲线和动态电位测试。电化学阻抗谱的测量是在开路电位下进行,扫描频率范围为104~10-2Hz。阻抗谱的解析采用ZSimpWin软件。
1.5表面分析
用JSM-6480型扫描电镜(SEM)观察涂层的形貌;用其附属的电子能谱仪(EDS)分析涂层表面的化学成分。
2结果分析
2.1阳极氧化膜的表面形貌和成分分析
图1(a)为AZ31镁合金不同电压下形成的氧化膜表面SEM形貌。当电压为80和90 V时,氧化膜表面的孔洞是小圆形的,80 V时氧化膜小孔的数量相对90 V时少些,而且小孔要小些;当电压升高至100 V时,孔隙面积增大,形状由小圆孔变为条状;电压升至110 V时,镁合金表面阳极氧化膜变得粗糙疏松。孔隙作为阳极氧化时生成气体的析出通道,与氧化过程中火花放电是对应的,要完全避免氧化膜的孔洞,几乎是不可能的[13]。

图1 不同电压下形成的阳极氧化膜
A.J.Zouzulin等认为随着阳极氧化膜击穿电压增大,膜厚的增加,阳极氧化膜的孔径增加[14]。在110 V电压下生成的氧化膜粗糙多孔或有突起,这是因为当阳极氧化膜生成以后,电压较高,将阳极氧化膜反复击穿,产生过烧,冷却后的熔融金属氧化物疏松,平整度变差。
图1(b)为AZ31镁合金在不同电压下阳极氧化后的EDS能谱图。从图1(b)可以看出,80,90和100 V时电压下氧化膜中O、Na、Mg、Si和K的含量相差不多, 其中Mg、O、和Si 3种元素的峰强最大,Na、K元素的峰强度较弱,Na、K可能是电解液残留在氧化膜中的元素。在电压增加到110 V时,氧化膜的成分和含量有较大变化,Na、K的含量明显增加,可能是110 V电压下的阳极氧化膜较疏松,因此表面吸附了较多电解液中的Na离子和K离子。
2.2极化曲线分析
图2为AZ31不同电压阳极氧化处理后的试样在Hank’s仿生溶液中的极化曲线,试样的自腐蚀电位Ecorr、腐蚀电流密度Icorr见表1。

图2镁合金及不同电压阳极氧化的试样在Hank’s溶液中的极化曲线
Fig 2 Polarization curves of the uncoated and MAO coated samples in Hank’s solution

表1 极化曲线所拟合的参数
从图2可以看出,80,90和100 V电压下生成的阳极氧化膜试样的极化曲线位置比镁合金的极化曲线位置向上移,腐蚀电位由-1.58 V升高到-1.48 V左右。而110 V电压下生成的阳极氧化膜试样的极化曲线位置比空白试样的曲线位置向下移,腐蚀电位由-1.58 V降低到-1.62 V左右。高电压下阳极氧化试样自腐蚀电位降低的结果在相关文献也有报道[15-17],研究认为[15]MgO和Hank’s溶液中的钙和磷酸根离子发生复合反应导致试样表面钝化性能和热力学稳定性能较差是导致阳极氧化试样试样自腐蚀电位降低的原因。从电流密度上看,阳极氧化试样在Hank’s仿生溶液的腐蚀电流密度均低于镁合金基体。从热力学的角度,试样的腐蚀电势正移的幅度越大,试样越不容易发生腐蚀。而电流密度的大小说明试样发生腐蚀后,腐蚀速率的快慢,电流密度越小,腐蚀速率越小。因此80,90和100 V电压下生成的氧化膜涂层有效提高了镁合金的耐蚀性,然而由表1可见3种条件下制备的阳极氧化试样的Ecorr和Icorr相差不大。
2.3阻抗谱分析
图3(a)和(b)分别为阳极氧化前后AZ31镁合金在Hank’s溶液中的Nyquist和Bode图。
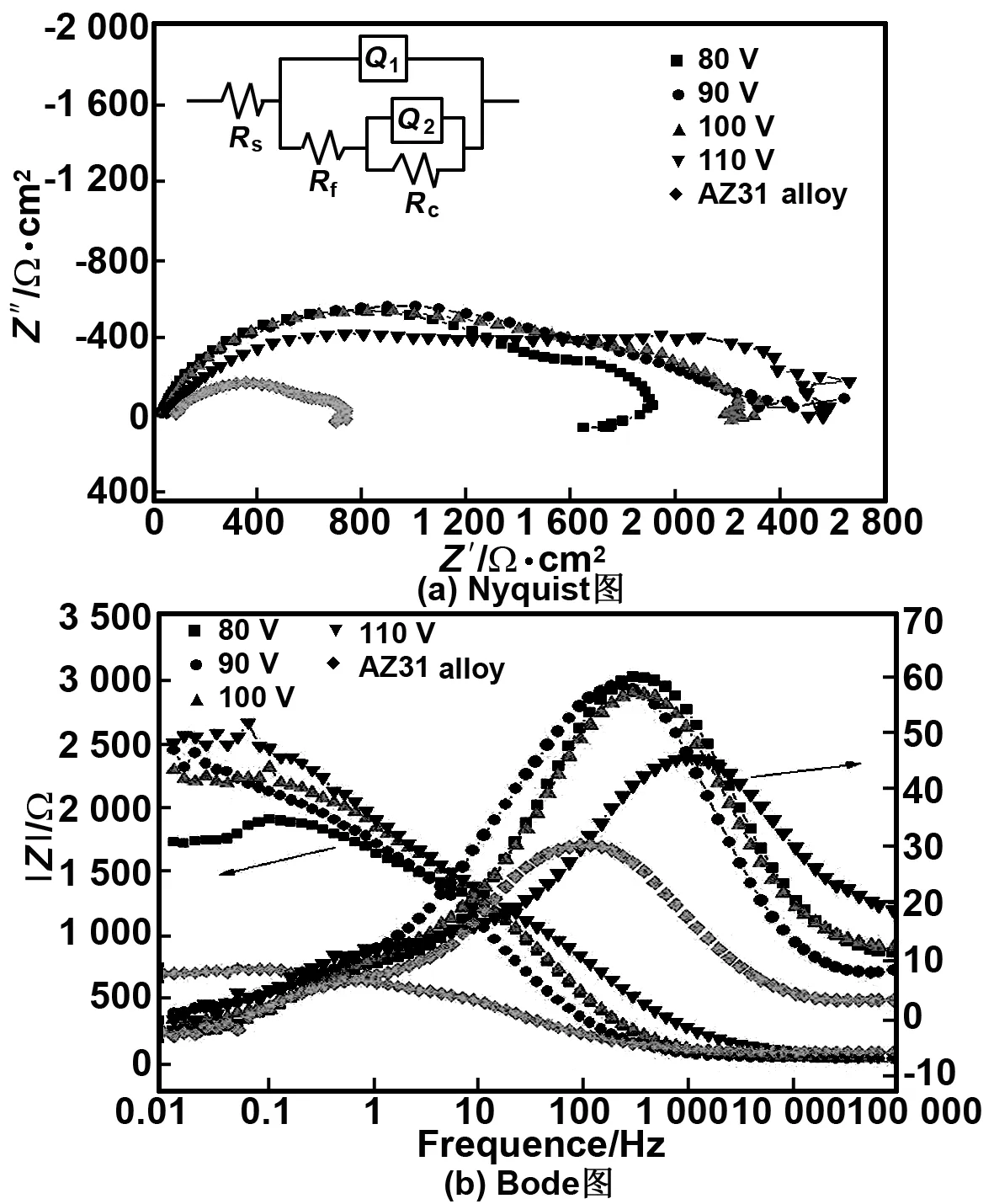
图3镁合金及不同电压阳极氧化的试样在Hank’s溶液中的EIS谱
Fig 3 EIS data of AZ31 alloy and MAO samples under different voltage in Hank’s solution
根据以上的EIS图谱,应用ZsimpWin软件所拟合的等效电路如图3(a),其等效电路由溶液电阻Rs、最外层膜表面/介质界面的常相位角元件Q1、最外层钝化层电阻Rf、阳极氧化层/金属基体界面的双电层电容Q2、电荷转移极化电阻Rct组成,相应的拟合数据如表2所示。由图3(a)可见,所有试样Nyquist图中的高频和中频端各有一个容抗弧。高频端反映的是氧化膜层信息,容抗弧的大小表示膜层的介电性能与屏蔽性能,中频端体现镁合金与溶液界面反应的信息,容抗弧的大小反映了金属腐蚀过程的界面双电层电容和电荷转移电阻。图3(a)阳极氧化试样的容抗弧均大于镁合金试样,表明阳极氧化试样具有较大的阻抗。由图3(b)的bode图可见,阳极氧化后的试样的相位角均高于镁合金,阻抗模值明显高于镁合金基体,说明阳极氧化层具有较低的电容,较高的电阻。其中80 V电压下的阳极氧化试样的阻抗模值低于其它阳极氧化试样。在Hank’s溶液中浸泡初期,AZ31镁合金表面形成新的钝化膜(氧化镁或氢氧化镁)[18]。由于钝化膜疏松,故浸泡10 min时,电解液渗透至金属和氧化膜中间形成双电层,因此在电化学阻抗谱等效电路中表现为双电层电容Q2。由表2拟合数据可见,100 V下的阳极氧化试样具有最大的涂层电阻(Rf=1 556 Ω·cm2),90 V下阳极氧化试样具有最大电荷转移电阻(Rct=1 258 Ω·cm2和极化电阻(Rp=2 469 Ω·cm2)。由于阳极氧化层表面存在大量的孔隙,微裂纹等离子导电通道,影响了氧化层的屏蔽功能,然而粗糙多孔的表层氧化膜可能在溶液中发生“自精饰”作用(根据表面化学原理,表层氧化膜的局部微小颗粒可能溶解,其产物进入氧化膜的小孔内),从而降低了腐蚀离子的渗透速率,导致膜电阻Rf的增加,提高了镁合金的耐蚀性[15]。

表2 根据等效电路所拟合的电化学参数
2.4析氢结果及分析
通常镁及其合金在溶液中与水发生化学反应生成Mg(OH)2和氢气,测量生成氢气的体积可得到镁腐蚀速率有关信息。
图4为AZ31镁合金基体和阳极氧化层试样在Hank’s仿生溶液中的析氢实验结果。从图4可以看出,浸泡前7 d,阳极氧化处理后的试样在Hank’s溶液中的析氢量不超过5 mL,析氢速率明显低于镁合金,随后析氢速率有所增高,表明阳极氧化可延缓镁合金降解。随后,阳极氧化试样的析氢速率有所增加。浸泡15 d后,镁合金析出氢气41 mL,阳极氧化试样的氢气析出量为14~21 mL,其中80,90和100 V 阳极氧化的试样析氢量变化不大,但110 V析氢量明显高于其它阳极氧化的镁合金试样,镁基体腐蚀程度较深,表明110 V下生成的氧化膜的耐腐蚀性能较差。
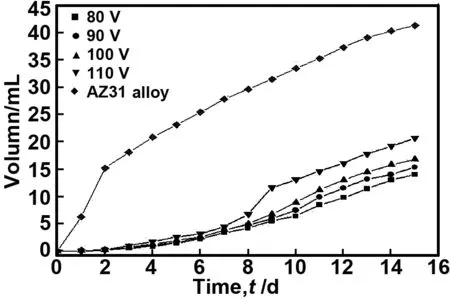
图4AZ31合金及阳极氧化处理后在Hank’s溶液中的析氢体积
Fig 4 Hydrogen evolution volume in Hank’s solution of the AZ31 alloy and MAO samples
图5为镁合金及不同电压阳极氧化处理后的试样在Hank’s溶液中浸泡15 d的微观形貌。表3为图5所示相应区域的成分分析。

图5 Hank’s溶液浸泡15 d的镁合金及阳极氧化后表面SEM形貌和EDS分析区域
Fig 5 SEM images and EDS spectrum areas of the AZ31 alloy and MAO samples under different voltage after immersion in Hank’s solution for 15 d
表3图5所示区域化学成分分析
Tablet 3 Chemical composition of the areas marked in Fig 5

Element/at%Area OMgAlPCaSiNa图5(a)Spectrum1Spectrum264.0970.4620.469.981.990.987.679.605.798.99图5(b)Spectrum1Spectrum268.0865.8418.1117.194.385.603.895.164.213.462.102.00图5(c)Spectrum1Spectrum269.0562.8412.5723.466.024.865.463.546.905.30图5(d)Spectrum1Spectrum271.1262.5315.7222.146.195.085.973.611.004.292.35图5(e)Spectrum1Spectrum266.9471.3910.2712.017.903.907.043.067.858.950.69
由表3浸泡后的成分变化可知,镁合金在浸泡过程中除腐蚀产生的氧化镁外,表面还检测出较多的P、Ca,说明Hank’s溶液中的磷酸根和钙离子参与了腐蚀反应,并沉积在镁合金表面形成磷酸钙盐的沉积,其中白色凸出部分(图5(a) spectrum 2)的P、Ca的含量高于黑色部分(图5(a) spectrum 1),试样表面裂纹是干燥脱水后所形成。阳极氧化试样在Hank’s溶液浸泡后,氧化层表面的孔隙消失,形成龟裂的表面,表面有白色沉积,如图5(b) spectrum 2,图5(c) spectrum 1, 图5(d) spectrum 1和图5(e) spectrum 2。表3可见,浸泡前后阳极氧化试样表面的O含量增加,Mg含量有所减少,并同样检测出较多的P和Ca元素,其中80,90和100 V电压下的阳极氧化试样表面的白色沉积处的P和Ca的含量高于表面的其它位置,110 V电压下阳极氧化试样的白色凸起处(图5(e) spectrum 2)P和Ca的含量则较低,Si含量较高。表明80,90和100 V电压下阳极氧化试样多孔的表层在溶液中发生局部微小颗粒的溶解,且Hank’s溶液中的磷酸根和钙离子参与了溶解过程,其产物进入氧化膜的小孔内,并在表面形成磷酸钙盐的沉积,形成无孔表面。而110 V电压下的阳极氧化试样由于凹凸不平,因此浸泡后仍较不平整,凸起处除少量的磷酸钙盐的沉积外,Mg和Si的含量与浸泡前的相近,而凹陷处有较多的磷酸钙沉积。
3结论
(1)随阳极氧化电压增大,阳极氧化膜表面的孔隙增大,电压升至110 V时,氧化膜反复击穿,产生过烧,冷却后的熔融金属氧化物疏松,平整度较差。
(2)阳极氧化可提高镁合金在Hank’s仿生溶液的耐蚀性,延缓镁合金降解1周左右。其中80,90和100 V电压下生成的阳极氧化膜试样的耐蚀性相差不大。而110 V电压下生成的阳极氧化膜试样的腐蚀电位降低,析氢量明显高于其它试样,耐蚀性低于其它阳极氧化试样。100 V下的阳极氧化试样具有最大的涂层电阻,90 V下阳极氧化试样具有最大电荷转移电阻和极化电阻。
(3)浸泡过程中,Hank’s溶液中的磷酸根和钙离子参与了溶解过程,其产物进入氧化膜的小孔内,并在表面形成磷酸钙盐的沉积,形成无孔表面。
参考文献:
[1]Xin Y, Hu T, Chu P K. In vitro studies of biomedical magnesium alloys in a simulated physiological environment: a review [J]. Acta Biomaterialia, 2011, 7 (4): 1452-1459.
[2]Zeng R C, Dietzel W, Witte F, et al. Progress and challenge for magnesium alloys as biomaterials [J]. Adv Eng Mater, 2008, 10: B03-B14.
[3]Renato A A, Mara C L. Corrosion fatigue of biomedical metallic alloys:mechanisms and mitigation [J]. Acta Biomaterialia, 2012, 8(5): 937-962.
[4]Witte F, Hort N, Vogt C,et al. Degradable biomaterials based on magnesium corrosion [J]. Current Opinion in Solid State and Materials Science, 2008, 12(5): 63-72.
[5]Witte F, Fisher J, Nellesen J,et al. In vitro and in vivo corrosion measurements of magnesium alloys [J]. Biomaterials, 2006, 27(7): 1013-1018.
[6]Hong Yansong,Yang Ke,Zhang Guangdao, et al.The role of bone induction of a biodegradable magnesium alloy [J]. Acta Metallurgica Sinica, 2008, 44(9):10351341.
洪岩松, 杨柯, 张广道,等. 可降解镁合金动物体内骨诱导作用[J]. 金属学报, 2008, 44(9): 1035-1041.
[7]Zeng Rongchang, Kong Linhong, Chen Jun, et al.Research progress on surface modification of magnesium alloys for medical applications [J]. The Chinese Journal of Nonferrous Metals, 2011, 21(1): 35-43.
曾荣昌, 孔令鸿, 陈君,等. 医用镁合金表面改性研究进展[J]. 中国有色金属学报, 2011, 21(1): 35-43.
[8]Song G L. Control of biodegradation of biocompatable magnesium alloys [J]. Corrosion Science, 2007, 49(4): 1696-1701.
[9]Pan Y K, Chen C Z, Wang D G,et al. Preparation and bioactivity of micro-arc oxidized calcium phosphate coatings [J]. Materials Chemistry and Physics, 2013,141 (2-3) : 842-849.
[10]Zhang X P, Zhao Z P, Wu F M, et al. Corrosion and wear resistance of AZ91D magnesium alloy with and without microarc oxidation coating in Hank’s solution [J]. Journal of Materials Science, 2007, 42(20): 8523-8528.
[11]Guo Lei, Liu Kui, Zhang Shiliang, et al.Cytotoxicity of AZ31B magnesium alloy covering with magnesium oxide [J]. Rare Metal Materials and Engineering,2008, 37(6): 1027-1031.
郭磊, 刘魁, 张世亮,等. 氧化镁膜AZ31B镁合金材料的细胞毒性研究[J]. 稀有金属材料与工程, 2008, 37(6): 1027-1031.
[12]Yang X M, Li M, Lin X,et al. Enhanced in vitro biocompatibility/bioactivity of biodegradable Mg-Zn-Zr alloy by micro-arc oxidation coating contained Mg2SiO4[J]. Surface & Coatings Technology, 2013, 233(10): 65-73.
[13]Zhang Y J, Yan C W, Wang F H, et al. Study on the environmentally friendly anodizing of AZ91D magnesium alloys [J]. Surface and Coatings Technology, 2002, 161(1): 37-44.
[14]Zozulin A J, Bartak D E. Anodized coatings for magnesium alloys [J]. Metal Finishing, 1994, 92(3): 39-44.
[15]Gan J J, Tan L L, Yang K, et al. Bioactive Ca-P coating with self-sealing structure on pure magnesium [J]. J Mater Sci: Mater Med, 2013, 24 (2):889-901.
[16]Liang J, Srinivasan P B, Blawert C,et al. Electrochemical corrosion behaviour of plasma electrolytic oxidation coatings on AM50 magnesium alloy formed in silicate and phosphate based electrolytes[J]. Electrochim Acta, 2009, 54(1): 3842-3850.
[17]Srinivasan P B, Liang J, Blawert C, et al. Characterization of calcium containing plasma electrolytic oxidation coatings on AM50 magnesium alloy [J]. Appl Surf Sci, 2010, 256(12): 4017-4022.
[18]Zhu Y Y, Wu G M, Zhang Y H, et al. Growth and characterization of Mg(OH)2film on magnesium alloy AZ31 [J]. Applied Surface Science,2011, 257(14): 6129-6133.
A study on the effects of degradation performance of magnesium alloy in Hank’s solution by anodization treatment
ZHANG Chunyan,LIN Panni,YANG Mingbo,XU Chaoling
(School of Materials Science & Engineering, Chongqing University of Technology, Chongqing 400054,China)
Abstract:The degradable performances of magnesium alloy in Hank’s solution after anodization treatment at different voltage were measured by electrochemical measurement technology and immersion test.The morphologies and chemical component of the anode oxide film before and after immersion in Hank’s solution were analyzed by SEM and EDS to discuss the degradation behavior in simulated body fluid. The results indicate that the average size of the pores on the oxide films were increased with the applied voltage, the film ablated with loosen and uneven surface when the voltage reached to 110 V. Anodize oxidation treatment increased the corrosion resistance of Mg substrate signi�cantly. Sample at 100 V anodization treatment exhibited largest coating resistance (Rf) and 90 V anodized sample has the highest charge transfer resistance (Rt) and polarization resistances (Rp). The anodized samples could postpone the degradation of magnesium alloy substrate about one week’s in Hank’s solution, whereafter the hydrogen evolution rates somewhat increased. The corrosion potential (Ecorr) of the 110V anodized sample was more negative than the Mg substrate and the hydrogen evolution rates was higher than that of other coated samples after one week’s immersion. During the immersion, calcium phosphate was formed with the slow dissolution of anode oxide layer due to theanddeposition on the coated surface. The reaction products entered the pores of the coating and formed the non-porous surfaces.
Key words:magnesium alloys;anodized oxidation;degradation performance;biomaterials
文章编号:1001-9731(2016)06-06119-06
* 基金项目:国家自然科学基金资助项目(51201192)
作者简介:张春艳(1974-),女,吉林通化人,副教授,博士,硕士生导师,主要从事医用镁合金表面改性及腐蚀性能的研究。
中图分类号:TG146.4
文献标识码:A
DOI:10.3969/j.issn.1001-9731.2016.06.021
收到初稿日期:2015-05-12 收到修改稿日期:2015-10-09 通讯作者:张春艳,E-mail:zhangchunyan@cqut.edu.cn
猜你喜欢
表面工程与再制造(2022年1期)2022-05-25 13:22:10
重型机械(2020年3期)2020-08-24 08:31:40
科学中国人(2017年35期)2017-06-08 06:12:33
电镀与环保(2017年2期)2017-05-17 03:42:19
电镀与环保(2017年1期)2017-02-27 08:02:25
电镀与环保(2016年2期)2017-01-20 08:15:25
电镀与环保(2016年2期)2017-01-20 08:15:23
当代化工研究(2016年6期)2016-03-20 16:21:48
焊接(2016年1期)2016-02-27 12:57:21
电镀与环保(2015年5期)2015-03-11 15:29:40
