
Theoretical Investigation of the Electronic Structure and Optical Properties of CuSin and Clusters (n=4~10)
2016-07-12 12:57:57LINLinYANGJucai
光谱学与光谱分析 2016年9期
LIN Lin, YANG Ju-cai
1.College of Science, Inner Mongolia University of Technology, Huhhot 010051, China 2.School of Chemical Engineering, Inner Mongolia University of Technology, Huhhot 010051, China 3.School of Energy and Power Engineering, Inner Mongolia University of Technology, Huhhot, 010051, China
LIN Lin1,2, YANG Ju-cai2,3*
1.College of Science, Inner Mongolia University of Technology, Huhhot 010051, China 2.School of Chemical Engineering, Inner Mongolia University of Technology, Huhhot 010051, China 3.School of Energy and Power Engineering, Inner Mongolia University of Technology, Huhhot, 010051, China
The electronic structure and UV-Vis properties of ground state CuSin(n=4~10) and CuSinanion clusters were studied using B3LYP density functional theory (DFT) at a 6-311+G (d) level.Calculations indicate that: (1) the band gap of neutral CuSinclusters is narrower than their anion, indicating anion clusters are relatively stable; (2) the energy gap and electronic structure calculations indicate that the anion CuSi5cluster is more stable than neighboring clusters; and (3) the UV-Vis spectrum of CuSinclusters and CuSinanions suggests that the neutral clusters are weakly absorbing; the anion clusters are strongly absorbing, and anion clusters with increasing size of the Si atoms experience a redshift in the absorption spectra.
Copper doped silicon clusters; Electronic structure; Absorption spectrum
Introduction
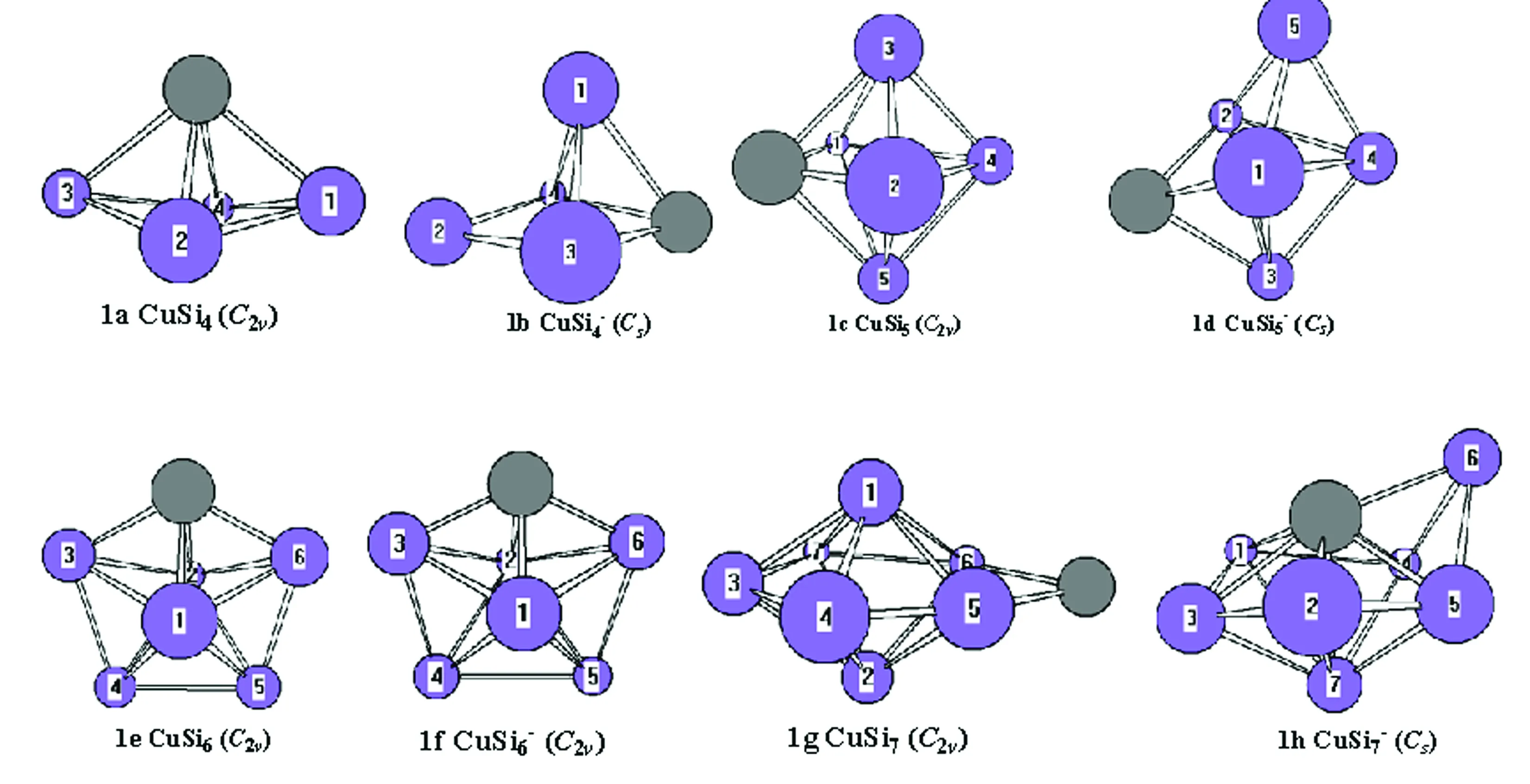
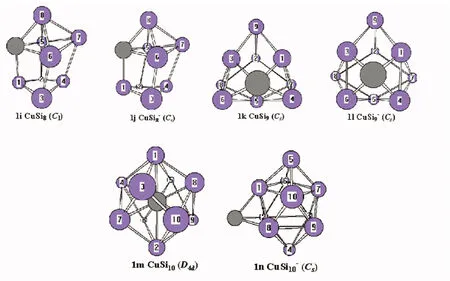
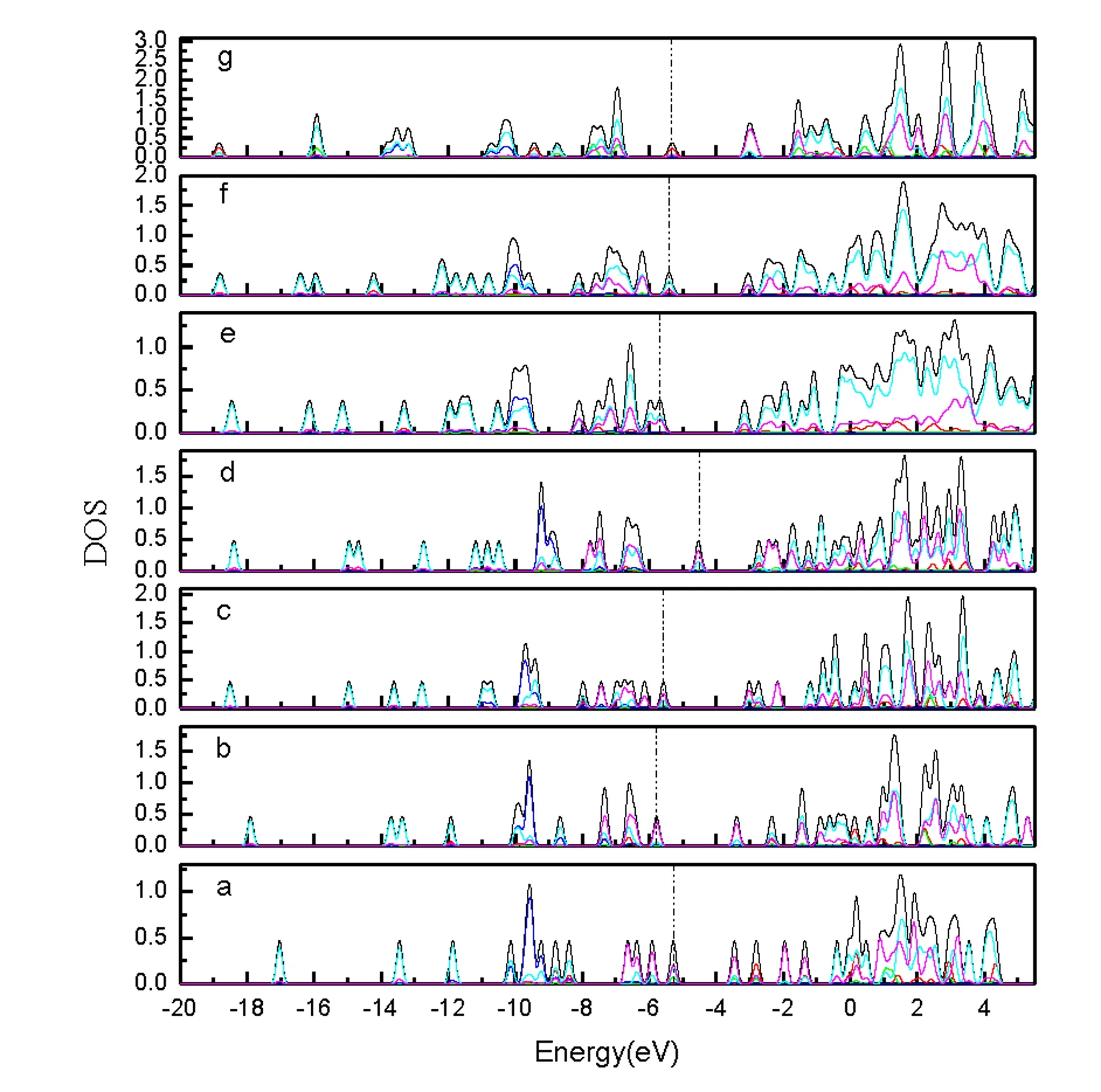
Silicon atoms (Si) and nanoclusters have drawn more and more attention because of their relevance in the development of nanelectronics[1-3].Pure silicon clusters are unstable due to the “dangling bonds”on their surfaces[4-6]but the process of impurity-doping is used to stabilize pure silicon clusters.Transition metal (TM) atoms are considered to be good candidates for doping, as they have unfilled d-orbitals containing single-electrons, which can effectively saturate the “dangling bonds" of the silicon clusters[7-9].After the TM is added into the silicon frame, the clusters tend to form closed shell electronic structures and generate transitional metal encapsulated clusters with novel properties.Beck[10, 11]first observed the stable metal-doped silicon clusters in a supersonic molecular beam coupled with a mass spectrometer.The dominant clusters in this study were silicon clusters with one metal atom attached:MSin(M=Cr, Mo, and W).Hiura et al.[12]studiedMSin,M=HF, Ta, W, Re, etc ….(8 Copper (Cu) is a common trace impurity in the manufacturing of silicon devices, such as integrated circuits and solar cells.Since copper plays an important role in altering the electronic properties of Si-based semiconductors, copper, behavior in Si has been studied extensively.Xiao et al.[15]performed a comparative study of the interaction ofM(M=Scandium (Sc) and Cu) atoms with Sin(n=1~6) clusters using a B3LYP/6-311+G(d) method, in which the most stable isomers of ScSin(n=1~6), binding energies, vertical and adiabatic ionization potentials, and electron affinities were reported. In Sec.Ⅱ, we provide the computational details.In Secs.Ⅲ, we discuss electronic structure and ultraviolet-visible spectroscopy of the ground-state structures CuSin(n=4~10) clusters and their anions, respectively.Finally, we summarize our results in Sec.Ⅳ. The electronic properties of CuSinclusters were examined using the DFT at a B3LYP/6-311+G (d) level.We chose the B3LYP approach, because this method has been previously by Xiao et al.[16], Hossain et al.[18], and Dkhissi et al.[22]and B3LYP with either the 6-31+G* or 6-311+G* basis set is well suited for investigating the electronic structural properties of CuSinclusters. The ultraviolet-visible (UV-Vis) spectra was calculated using a time-dependent B3LYP method based on the optimized structure.Time-dependent DFT(TD-DFT)[23-25]has proven to be a powerful and effective computational tool for the study of ground and excited state properties. The ground state structure we used is from the literature[26]and is shown in Fig.1.To characterize the relative stabilities of all CuSinclusters considered, the gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were considered useful quality to evaluate the chemical stability.A larger energy gap often corresponds to a higher stability.In this paper, the HOMO-LUMO gap (Egap) was calculated as follows Egap=E(LUMO)-E(HOMO) AllthecalculationswerecarriedoutusingGaussian09package[27]andMultiwfnprogram,revision2.1[28-29]. Fig.1 The isomers of CuSin (n=4~10) and their anions are obtained at the MP2/6-31G(2df,p)level.Only silicon atoms are numbered In the following, we will describe the electronic structures and optical properties of individual CuSin(n=4~10) clusters and their anions. 2.1 Electronic structure properties Fig.3 The total density of state (TDOS) and partial density of state (PDOS) map of CuSin(n=4~10), The DOS value is in arbitrary unit.The vertical broken line shows the HOMO.Gaussian function with full width half maximum (FWHM) of 0.25 eV was used for broadeningmolecular orbital energies to yield the DOS curves.Black represents TDOS, red represents Cuatomic s orbital, green represents Cu atomic p orbital, blue represents Cu atomic d orbital, cyanrepresents Si atomic s orbital and pink represents Si atomic p orbital 2.2 Ultraviolet-Visible spectroscopy Fig.5 The absorption spectra of CuSin(n=4~10) clusters (a)—(g) Fig.6 The absorption spectra of clusters (a)—(g) In this paper, The electronic structure of the neutral CuSin(n=4~10) clusters and their anions was examined using B3LYP/6-311+G (d) level, UV-Vis spectroscopy of the neutral CuSin(n=4~10) clusters and their anion were examined TD-B3LYP/6-311+G (d) level The results are summarized below: (2) The results showed that both the neutral CuSin(n=4~10) clusters and their anion clusters have obvious energy gaps.Anion clustern=5 andn=10 have large band gaps, which suggests that they have higher stability than their neighboring clusters. (3) The UV-Vis spectroscopy suggestes that the neutral clusters have weak absorption, while their anion clusters have strong absorption.Anion clusters (exceptn=5) with the increase of the Si atomic size there is redshift in absorption spectra. [1] Jarrold M F.Science,1991, 252:1085. [2] Brown W L,Freeman R R,Raghavachari K,et al.Science,1987, 235:860. [3] Hayashi S,Kanzawa Y,Kataoka M,et al.Phys.D Atom.Mol.Cl., 1993, 26: 144. [4] Zhu X L, Zeng X C.J.Chem.Phys., 2003, 118: 3558. [5] Kaxiras E.Phys.Rev.Lett., 1990, 64: 551. [6] Kaxiras E, Jackson K.Phys.Rev.Lett., 1993, 71: 727. [7] Majumder C, Kulshreshtha S K.Phys.Rev.B, 2004, 70: 245426. [8] Zorriasatein S, Joshi K, Kanhere D G.Phys.Rev.B, 2007, 75: 045117. [9] Wang J, Ma QM, Xie Z, et al.Phys.Rev.B, 2007, 76: 035406. [10] Beck S M.J.Chem.Phys.,1987,87:4233. [11] Beck S M.J.Chem.Phys., 1989, 90: 6306. [12] Hiura H,Miyazaki T,Kanayama T.Phys.Rev.Lett., 2001, 86: 1733. [13] Khanna S N, Rao B K, Jena P.Phys.Rev.Lett., 2002, 89: 016803. [14] Jackson K, Nellermoe B.Chem.Phys.Lett., 1996, 254: 249. [15] Xiao C Y, Abraham A, Quinn R, et al.J.Phys.Chem.A, 2002, 106: 11380. [16] Xiao C Y, Hagelberg F, Lester W A.Phys.Rev.B, 2002, 66: 075425. [17] Guo L J, Zhao G F, Gu Y Z, et al.Phys.Rev.B, 2008, 77: 1954. [18] Hossain D, Pittman C U Jr, Gwaltney S R.Chem.Phys.Lett., 2008, 451: 93. [19] Lan Y Z, Feng Y L.Phys.Rev.A, 2009, 79: 033201. [20] Li G L, Ma W L, Gao A M, et al.Journal of Theoretical and Computational Chemistry, 2012,11:185. [21] Xu H G, Wu M M, Zhang Z G, et al.J.Chem.Phys.2012,136:104308. [22] Dkhissi A.Int.J.Quantum Chem., 2008, 108: 996. [23] Bader R F W.Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford (UK), 1990. [24] Bauernschmitt R, Ahlrichs R.Chem.Phys.Lett., 1996, 256: 454. [25] Kose E, Atac A, Karabacak M, et al.Spectrochim.Acta A,2012,97:435. [26] Lin Lin, Yang Jucai.J.Mol.Model.,Accepted. [27] Frisch M J, Trucks G W, Schlegel H B, et al.Revision C.01, Gaussian, Inc., Wallingford CT, 2010. [28] Tian Lu, Chen F.J.Comp.Chem., 2012, 33: 580. [29] Lu T, Multiwfn, Revision 2.1, University of Science and Technology Beijing, Beijing, China, 2011. *通讯联系人 O433.1 A 林 琳1,2, 杨桔材2,3* 1.内蒙古工业大学理学院,内蒙古 呼和浩特 010051 2.内蒙古工业大学化工学院,内蒙古 呼和浩特 010051 3.内蒙古工业大学能动学院,内蒙古 呼和浩特 010051 基于密度泛函的B3LYP/ 6-311+G (d)方法研究基态结构CuSin(n=4~10)和 CuSin阴离子团簇的电子结构和紫外吸收谱。计算结果表明:(1)中性CuSin团簇的带隙要比阴离子团簇的带隙要小,说明阴离子团簇比中性的要稳定;(2)阴离子CuSi5团簇要比相邻的其他团簇稳定;(3)紫外吸收谱可看出中性CuSin团簇属弱吸收而阴离子则表现出很强的吸收。对阴离子来说,随着硅原子的增加有红移现象发生。 铜掺杂硅团簇;电子结构;吸收谱 2015-09-16, 2016-01-12) Foundation item:the National Natural Science Foundation of China (21263010, 11562016) 10.3964/j.issn.1000-0593(2016)09-3026-07 Received:2015-09-16; accepted:2016-01-12 Biography:LIN Lin, (1974—), female, lecturer in College of Science, Inner Mongolia University of Technology e-mail: linlin@imut.edu.cn *Corresponding author e-mail: yangjc@imut.edu.cn
1 Computational Detalls
2 Results and Discussion
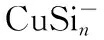
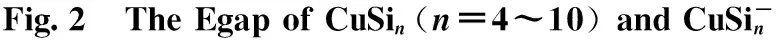
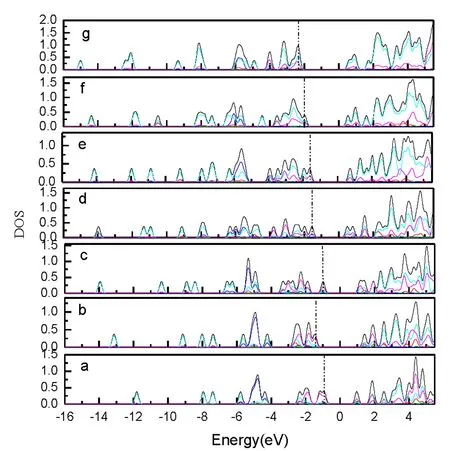
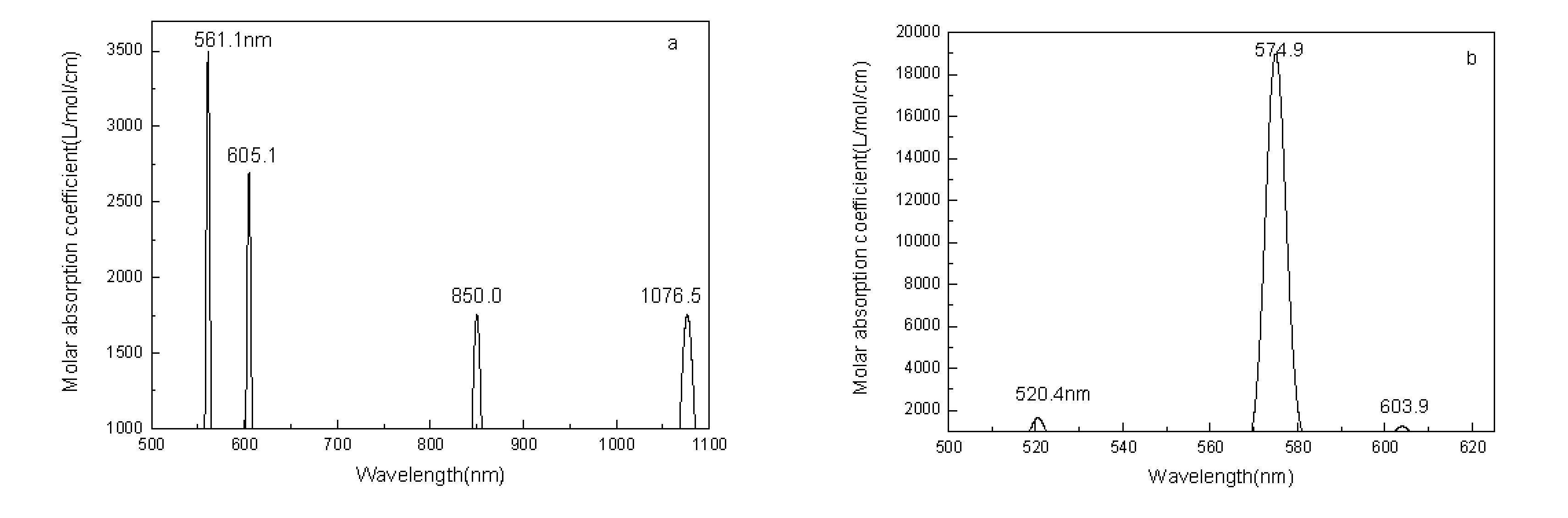

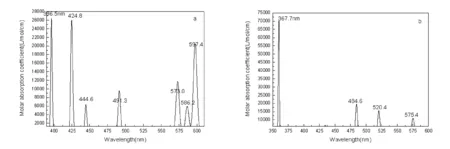
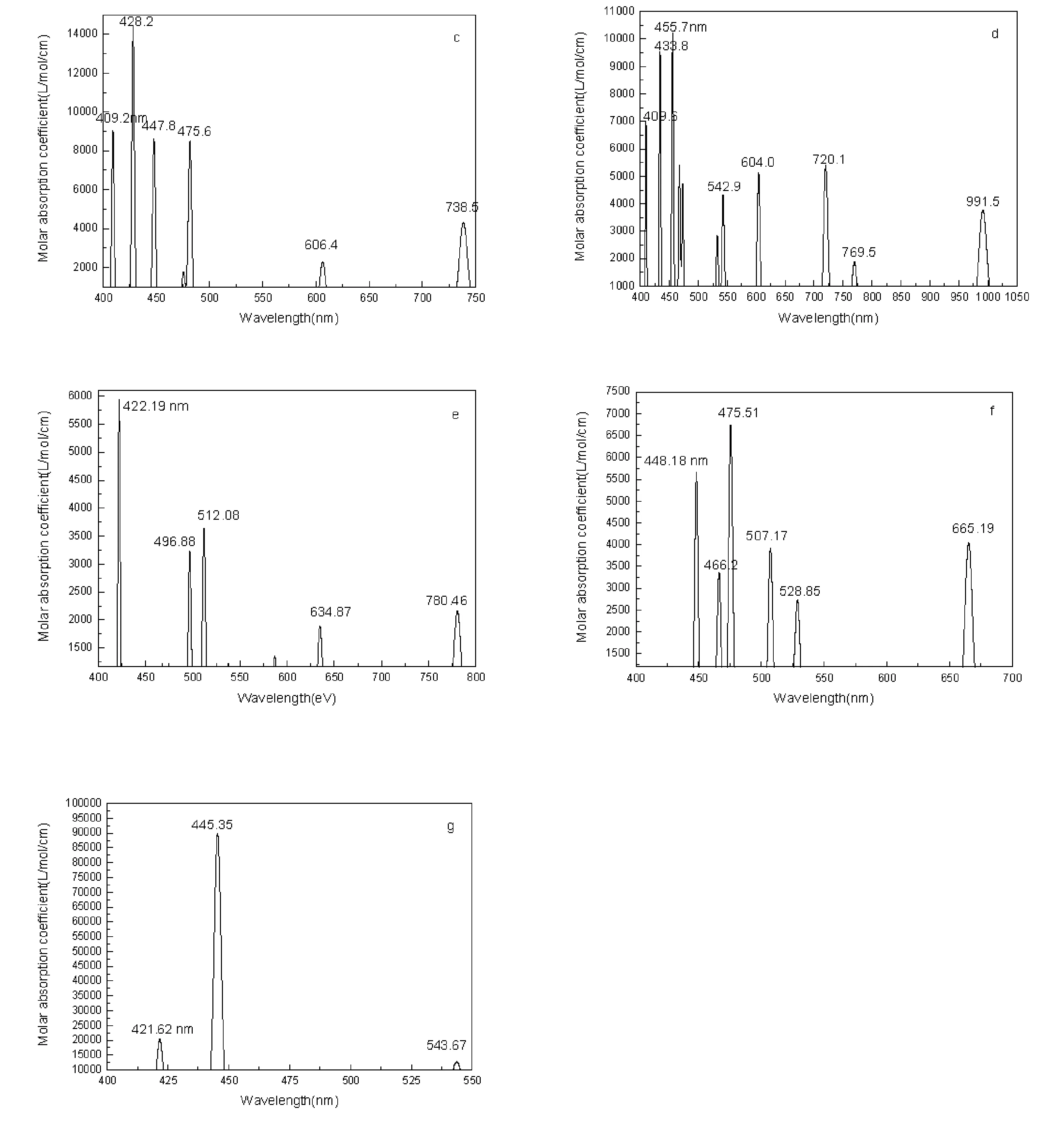
3 Conclusions
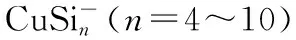
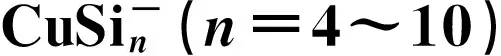
猜你喜欢
无机化学学报(2024年1期)2024-01-20 03:55:50
济南大学学报(自然科学版)(2021年6期)2021-11-10 01:25:42
化工职业技术教育(2021年5期)2021-11-09 03:10:40
儿童故事画报(2021年7期)2021-10-04 23:51:41
化工职业技术教育(2021年1期)2021-03-15 06:57:44
食品研究与开发(2020年18期)2020-09-10 03:34:04
成都信息工程大学学报(2019年3期)2019-09-25 08:31:14
振动工程学报(2017年4期)2018-05-31 12:38:00
电子制作(2018年1期)2018-04-04 01:48:38
草原歌声(2017年4期)2017-04-28 08:20:43
