
CRISPR/Cas9技术的发展及在基因组编辑中的应用
2016-06-23 13:49张凯丽李瑞胡桐桐徐永杰
生物技术通报 2016年5期
张凯丽 李瑞 胡桐桐 徐永杰
(信阳师范学院生命科学学院,信阳 464000)
技术与方法
CRISPR/Cas9技术的发展及在基因组编辑中的应用
张凯丽 李瑞 胡桐桐 徐永杰
(信阳师范学院生命科学学院,信阳 464000)
CRISPR/Cas9是由细菌和古细菌等微生物中特有的获得性免疫系统发展起来的基因组编辑技术,可以被一段短的RNA引导到复杂基因组中的特定位置,从而对靶标识别切割。该技术可以很容易对几乎所有生物体中的内源基因组DNA序列及其表达产物进行有选择地被编辑或调控,已成为一种热门的基因组编辑工具,正积极推动着从基础生物学到生物技术和医学等方面的发展。介绍CRISPR/Cas9的研究历史、结构和功能以及、精确识别的分子基础,并就其在基因组编辑中的应用进行了较为详尽的综述,以期为从事该领域的科研人员提供参考。
核酸内切酶;Cas9;PAM;CRIPR/Cas;基因组编辑
基因组编辑是指对基因组或表达产物(如转录物)进行针对性修饰的过程。真核生物基因组包含上亿万个碱基,做到精确操纵很困难,但在真核生物中特别是哺乳动物细胞中能够简单有效地做到,这对生物学基础理论、生物技术及医药领域等方面研究无疑都将起到巨大推动作用。目前,针对通过引入特定位点的DNA双链断裂来实现高效的基因编辑,已开发了4类DNA结合蛋白:来源于微生物移动遗传元件的巨核酶技术[1]、基于真核转录因子的锌指核酸酶(zinc finger nucleases,ZFN)[2]、来自黄单胞菌的真核转录激活因子样效应物(transcription activator-like effectors,TALEs)[3]以及来自II型细菌免疫系统——成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)由短RNA介导的DNA核酸内切酶Cas9[4,5]。巨核酶、ZFN和TALEN都是通过蛋白-DNA相互作用来识别特定的DNA序列。巨核酶整合了核酸酶和DNA结合结构域,而ZFN和TALEs蛋白由单个模板组成分别靶向DNA的3个或1个核苷酸,能够以所需的组合方式被任意组装,并连接上FokⅠ核酸酶结构域从而对特定基因位点切割。然而,这些技术平台都或多或少都存在缺限。巨核酶蛋白的残基及其靶向的DNA序列之间缺乏明确的对应关系,巨核酶作为一种基因编辑平台并没有被广泛接受。而ZFN结构域被装配成一个大的阵列时,由于相邻模块之间的干扰会造成背景依赖性的结合偏好[6],尽管已开发出许多策略来弥补这些缺陷,但ZFN的功能组件与DNA的特异性结合仍然是一个巨大的挑战[7]。同样的,尽管TALEs的DNA结合单体在大多数情况下是模块化的,但仍然可能因为特定的背景依赖性受到影响,而且所需的大量重复序列致使构建新的TALE阵列工作变得费力且昂贵[8]。
考虑到DNA结合蛋白在组装上的挑战,新的识别模式应能显著简化核酸内切酶的定制。CRISPR核酸酶 Cas9由一段短的gRNA(guide RNA)引导,依靠碱基互补配对识别靶DNA,几乎可以在所有生物体内有选择地直接编辑基因组DNA,从而在系统水平上揭示基因组的功能组成,并确定遗传变异的原因。目前,依靠Cas9的多重靶向也可以大规模地实现,它仅靠一连串短的gRNA介导而不是用大量的蛋白质组装。2015年,宾夕法尼亚州立大学的研究团队开发了从多顺反子基因生产大量gRNA的通用策略,大大提高了CRISPR的多重编辑效率[9]。因此,Cas9的快捷靶向以及其作为一种特定位点核酸酶的高效性和多重修饰的可能性,已经为从生物学基础研究到生物技术和医学应用研究开辟了一个广阔的领域,已在生物学研究上引发了继20世纪70年代DNA重组技术后又一场新的革命。本文主要就CRISPR/Cas9技术的发展及其在基因组编辑中的应用进行了综述。
1 CRISPR/Cas9的研究历史
有关Cas9核酸内切酶在基因组编辑中的应用,借鉴了过去10多年有关细菌和古细菌中神秘重复元件(现被称作CRISPR)的生物学功能研究成果(图1)。完整的II型CRISPR系统通常包括tracrRNA(反式激活嵌合RNA,trans-activating chimeric RNA)基因、Cas蛋白编码基因和CRISPR识别序列三部分(图2),tracrRNA位于5'端,一种非编码RNA,能够促进 crRNA的形成,也是Cas9蛋白发挥RNA介导的DNA切割作用必不可少的辅助因子;Cas蛋白编码基因位于中间,包括Cas9、Cas1、Cas2和Csn2;CRISPR识别序列位于3'端,由一个前导区(leader)、多个重复序列(repeat)和重复序列间的可变间隔序列(spacer)组成(图2),Spacer主要来源于噬菌体或质粒,包含21-72 bp碱基,不同的CRISPR基因座的间隔序列的数量差异较大,从几个到几百个不等。Cas基因被翻译成蛋白质,而大多数CRISPR序列首先被转录成一段单链RNA之后被加工成更短的CRISPR RNAs(crRNA),crRNA可以调控Cas酶的活性去降解靶核酸。
1.1 CRISPR的发现
关于CRISPR的研究最早可以追溯到1987年,Nakata等在研究大肠杆菌中Iap酶参与的碱性磷酸酶同工酶转换时发现,Iap基因下游有一段非常奇怪的29 nt重复序列[10]。该序列与其他重复元件不同的是这些序列 大多以串联重复的方式出现,其后 被5个32 nt的非重 复序列间隔开。但这个发现当时并没有引起足够重视,直到10多年后,随着越来越多的微生物基因组被测序,又陆续报道了来自不同细菌和古细菌菌株基因组的重复元件。这种重复元件在微生物基因组中可出现许多次,但每一重复元件都可以由不同的重复片段和间隔片段组成。Mojica等[11]把间隔重复序列分类,作为一个独特的家族,发现这些成簇的重复序列在超过40%的细菌和90%的古细菌中都存在,这也说明了CRISPR对细菌的重要性。
这些早期的发现激发了科研人员对微生物重复元件的兴趣,随后,Jansen[12]和Mojica[11]创造了用首字母缩写CRISPR来统一命名这些间隔重复序列[12,13]。与此同时,几组CRISPR相关基因也被确定是高度保守的,并且通常与重复元件相邻,这成为3种不同类型的CRISPR最终分类(类型I-III)的依据[14-16]。I型和III CRISPR系统包含多种Cas蛋白,形成crRNA复合物(CASCADE复合体为I型,CMR或CSM RAMP复合体为III型),以促进靶核酸的识别和切割,而II型系统Cas蛋白的数目却很少,只有Cas9(图2)。很快研究者就都把目光锁定在了II型系统上,因为依赖Cas9蛋白的CRISPR系统要比其他类型更简单。然而,尽管越来越多CRISPR位点被详细地描绘和注解,但其具体的生物学意义仍未阐明。
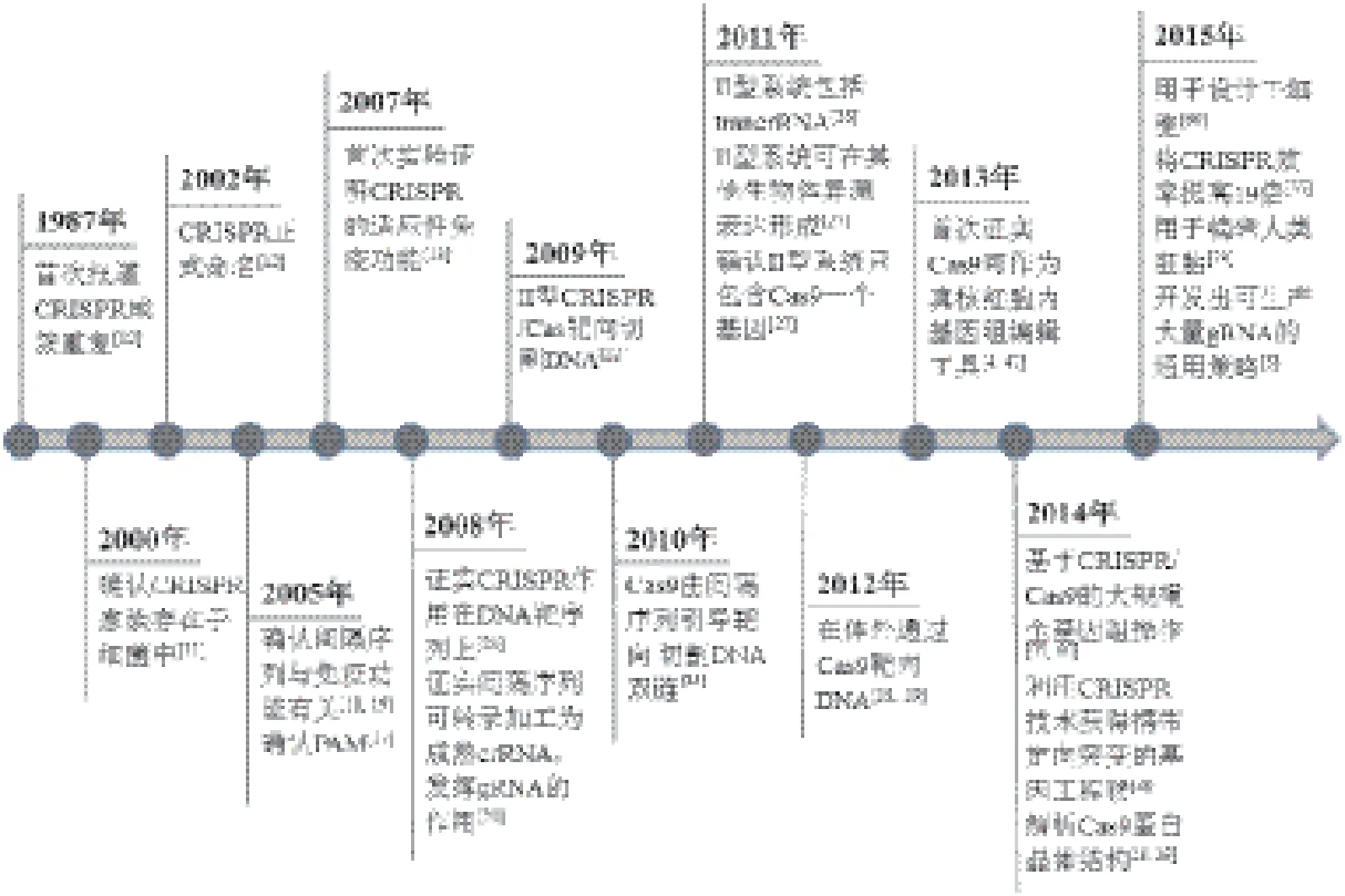
图1 CRISPR/Cas9技术发展历程
1.2 CRISPR的生物学意义
2005年,多个研究小组在对分离的重复间隔区序列进行系统分析时发现这些 CRISPR中的间隔DNA总是能够与噬菌体的DNA序列互补匹配[17-19]。这个发现是非常令人兴奋的,尤其是考虑到CRISPR基因座转录后病毒不能再感染携带有与病毒基因组一致的间隔序列的古细菌。同时,这些发现也引起了生物学家对CRISPR序列用作免疫记忆和防御机制的思考,并且个别间隔区可以通过利用核酸碱基互补配对来促进抵御噬菌体感染[18,19]。尽管这些显著的成就表明CRISPR位点可能参与了微生物免疫,但那些间隔序列如何介导病毒防御的特定机理仍不清楚。于是,美国马里兰州美国国家癌症生物技术信息中心的Eugene Koonin等提出了一个新的想法,即细菌和古细菌能够吸收噬菌体的DNA,然后将其留作己用,并转录出相应的RNA,与入侵的外源DNA结合,指导Cas酶裂解病毒DNA,这与真核生物采用的RNA干扰(RNAi)机制类似[17]。
2007年,丹麦的食品添加剂公司Danisco的Horvath等发现了II型CRISPR系统作为一个适应性免疫系统的实验证据:CRISPR间隔序列在以核酸为基础的免疫系统中指导特异性的靶向,而Cas酶控制间隔序列的获取和噬菌体防御[13]。随后,一系列研究阐明了CRISPR的防御机制,同时建立了3种类型CRISPR位点适应性免疫的机制。通过对大肠杆菌I型CRISPR基因座的研究,van der Oos等揭示了CRISPR序列被转录并转变成含有单个间隔序列的小crRNAs,并介导Cas核酸酶的活性[20]。同年,来自于表皮葡萄球菌的III-A型CRISPR系统中CRISPR介导的防御被证实可以阻碍质粒的结合,确立Cas酶的靶向活性是DNA而非RNA,后来在嗜热链球菌中发现不同的III-B型系统的研究显示crRNA指导的RNA也具有酶切活性[21,22]。
随着CRISPR的研究步伐推进,研究人员很快揭开每种类型CRISPR系统的许多细节。Bolotin等[17]证实间隔序列相邻基序(protospacer adjacent motif,PAM)可指导II型Cas9核酸酶切割DNA。Moineau等进一步明确了PAM序列的重要性,并揭示了PAM突变的噬菌体基因组可以有效避免被切割[23]。此外,在I型和II中,CRISPR序列内缺少PAM位点,有效避免了了CRISPR系统的自我靶向。2009年后,CRISPR系统的基本功能和机制已逐渐清晰,许多研究小组已开始将自然的CRISPR系统应用于各种生物技术,包括抗噬菌体乳酸菌的培养、细菌菌株的进化分类等[24]。然而,有关在基因组编辑的应用尚未被开发。
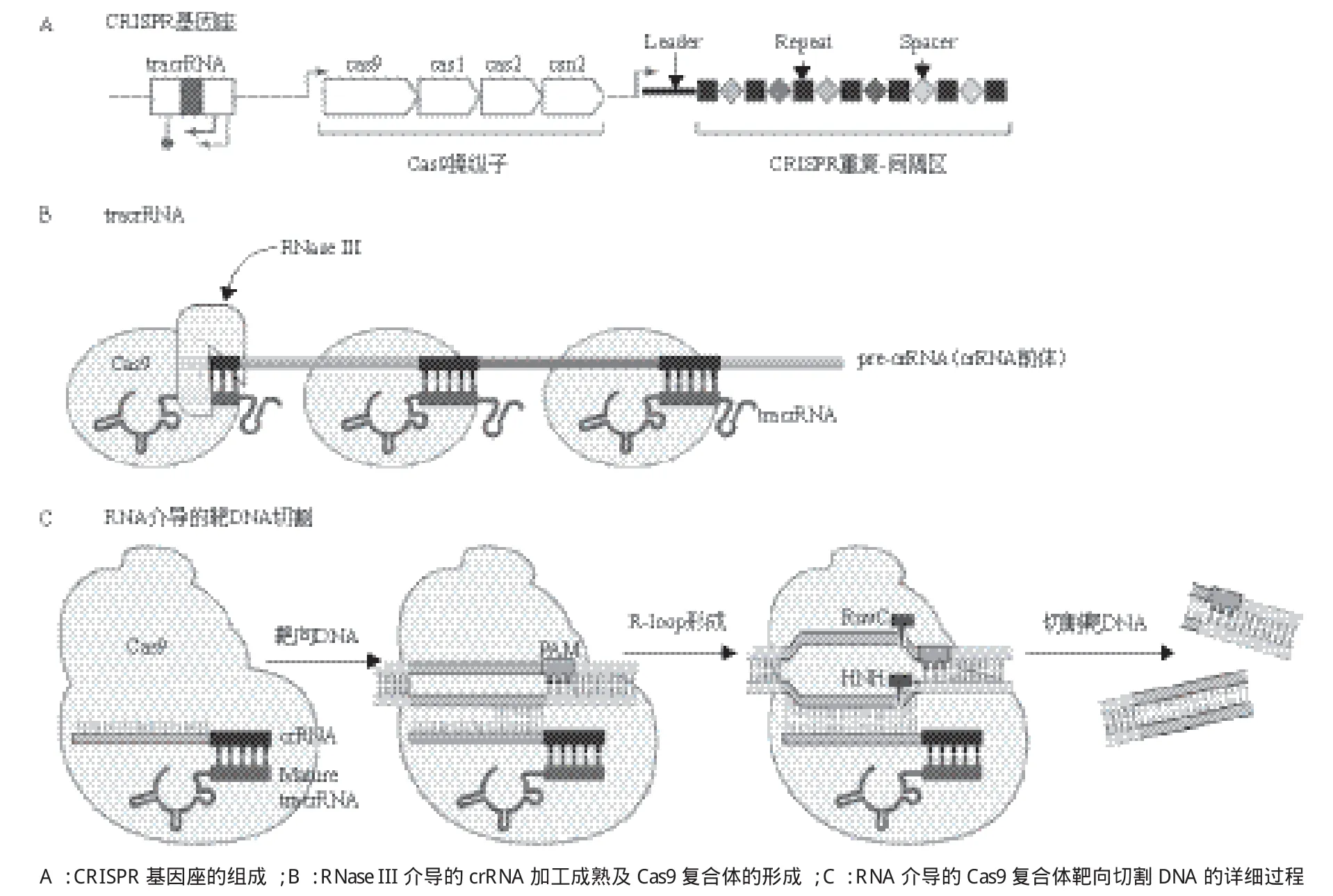
图2 II型CRISPR系统[30]
1.3 CRISPR/Cas9用于基因组编辑
2010年,两个研究II型CRISPR系统功能机理的小组发现设计一个简单可编辑的RNA对于基因组编辑是至关重要的。首先,Garneau和Moineau[25]及其同事利用遗传学研究嗜热链球菌证明了Cas9是Cas基因簇中唯一可以单独介导靶DNA裂解的酶。而Deltcheva和Charpentier等[26]发现II型CRISPR系统编码的tracrRNA可以指导RNaseⅢ和Cas9完成前体crRNA的成熟,而tracrRNA还可与成熟的crRNA的重复序列配对形成异源RNA二聚体,进而和Cas9结合成蛋白复合体,发挥识别和切割入侵的外源DNA的功能。这两项研究表明至少有3个组成部分(Cas9、成熟crRNA和tracrRNA)是对II型CRISPR核酸酶系统是必不可少的,这为将CRISPR用于基因组编辑奠定了基础。
2011年,Sapranauskas和Siksnys[27]和同事首先证明II型CRISPR系统是可转移的,将II型CRISPR基因座从嗜热链球菌移植到大肠杆菌中能够重建CRISPR干扰。到2012年,多个研究小组进一步作了生物化学鉴定,表明从嗜热链球菌或化脓性链球中纯化的Cas9可以通过crRNAs引导在体外切割靶DNA,这与之前的细菌研究结果是一致的[28,29]。此外,Jinek等[29]。提出了将tracrRNA和间隔RNA组合起来形成一个单链向导RNA(sgRNA,single-guide RNA)的想法,并且成功地构建出了sgRNA,将其与Cas9蛋白混合在一起,实现了对特定的DNA位点进行了切割这一重要发现,为进一步改造和利用CRISPR系统进行基因编辑奠定了坚实的基础。
2013年,两个研究小组首次报道了从嗜热链球菌和化脓性链球菌构建II型CRISPR/Cas9系统可用于哺乳动物细胞中的基因组编辑[4,5]。异源表达成熟的crRN A-tracrRNA杂交体以及sgRNAs指导Cas9切割哺乳动物细胞基因组可以激发内源性的NHEJ或HDR途径介导的DNA修复,从而实现基因组的定点编辑(图3),多种sgRNAs也可以一次性用于靶向多个基因[9]。从这些研究开始,Cas9逐渐受到研究者的青睐,被用于各种实验模型系统中的基因组编辑[8,30]。
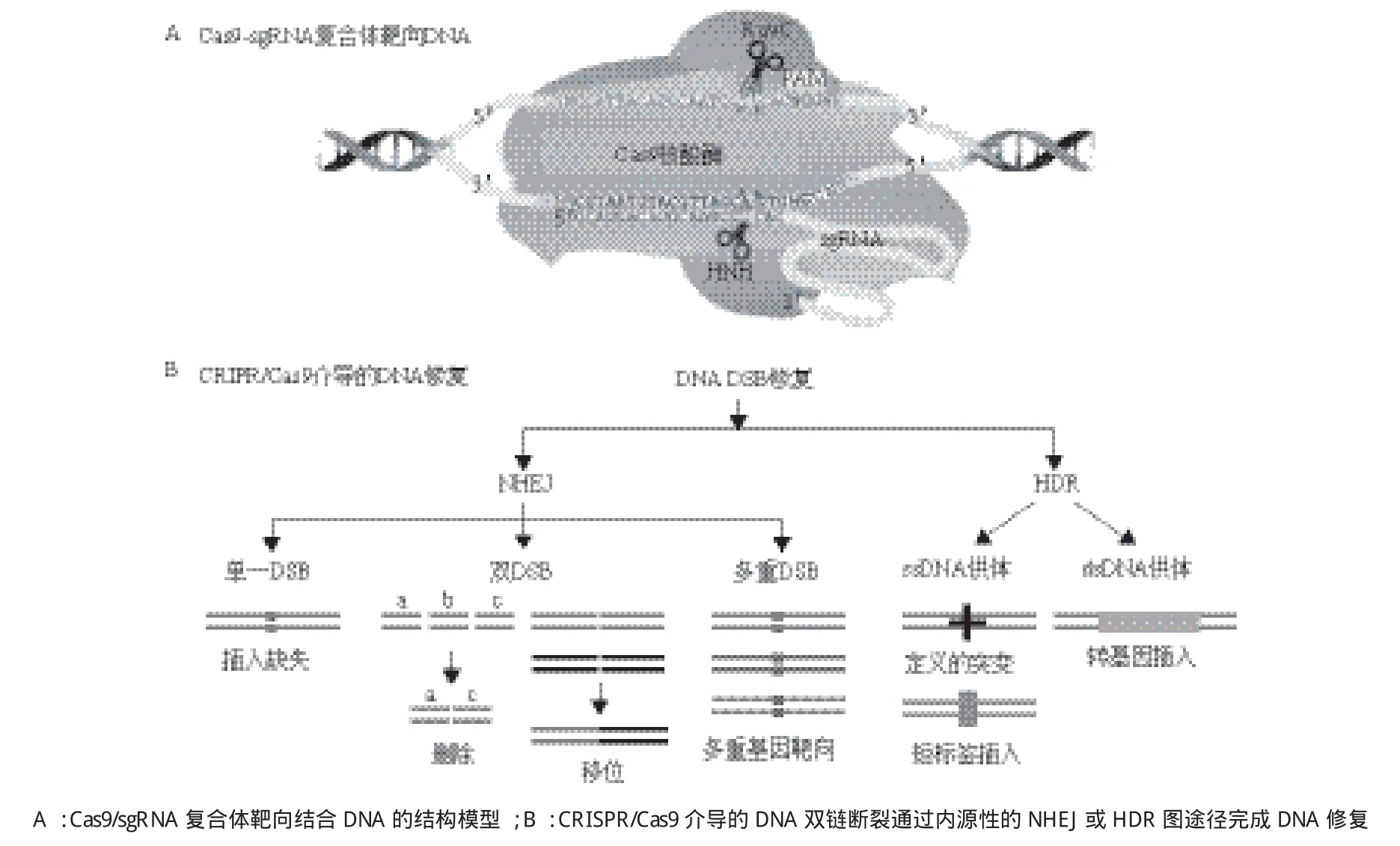
图3 CRISPR/Cas9介导的基因编辑[58]
2 Cas9的结构与功能的多样性
2.1 Cas9蛋白晶体结构
Cas9蛋白有两个核酸酶结构域——RuvC和HNH,HNH是一个单一的核酸酶结构域,而RuvC结构域却贯穿整个线性蛋白并被分成3个子结构域,RuvC I邻近Cas9蛋白N末端区域,RuvC II和III位于HNH结构域两侧临近蛋白的中间[31,32]。来自瑞士苏黎世大学和美国加州大学伯克利分校的研究人员利用X射线晶体分析法首次获得两种主要类型(链球菌和放线菌)的Cas9酶的三维晶体结构图,分辨率分别达到2.6埃和2.2埃分,然后利用单颗粒电子显微术揭示出Cas9与gRNA如何合作从而与靶DNA序列相互作用[32]。他们还发现尽管这两种Cas9酶催化区域外的结构有显著差异,但都具有相同的核心结构,该结构可以裂开两瓣形成钳状,当gRNA与Cas9结合后,可以激活Cas9活性。
此外,麻省理工的研究人员首次报道了Cas9复合体的高分辨率图像[33],发现Cas9主要分为两叶,即识别叶和核酸酶叶,其中识别叶参与识别gRNA和靶DNA组份,而核酸酶叶可分为Ruvc核酸酶结构域和HNH核酸酶结构域,前者主要结合切割靶基因的非互补链,而后者则结合切割互补链,导致双链断裂,破坏靶基因功能。他们还发现Cas9与gRNA之间的界面上的关键性结构,当Cas9准备切割靶DNA链时,这些结构允许Cas9在gRNA和靶DNA周围自我组装。而最近来自加州大学伯克利分校、德国马克斯普朗克生物物理化学研究所的研究人员解析了具有催化活性的化脓链球菌Cas9与85 nt sgRNA形成复合物的晶体结构,分辨率达到了2.9Å,由此揭示出了为识别靶DNA并预先组织形成一种Cas9-gRNA复合物的构象,这是一种不同于DNA结合状态的独特构象[34]。研究人员证实在该构象中识别DNA必需的10 nt的RNA“种子”序列呈现一种前序形态,而这一gRNA“种子区域”是启动识别DNA靶序列的基础。总的来说,Cas9晶体结构的揭示是CRISPR/Cas9技术有效用于基因组编辑技术的里程碑式发现,有望为科学家更好地完善该技术提供有力的支持。
2.2 Cas9蛋白的多样性
Cas9是II型CRISPR系统的主要特征,是一个大分子量的多功能蛋白,既能参与加工pre-crRNA产生成熟的crRNA,也能切割降解外源靶DNA。Cas9蛋白具有丰富的多样性,目前从同源序列数据库已鉴定出超过1 000个Cas9核酸酶,这些蛋白质长度变异较大,单大致在900-1 600个氨基酸,大多数Cas9蛋白的长度分布可分为两个分别以1 100和1 350个氨基酸为中心的群体[31]。
尽管蛋白质长度有明显差异,但是所有Cas9蛋白有着相似的结构域,包括RuvC和HNH核酸酶结构域以及REC域,还有一个螺旋富集区与富含精氨酸的螺旋桥[35,36]。不同于可在细菌和古细菌中存在的I型和III CRISPR系统,II型CRISPR系统迄今只存在于细菌中,大多数Cas9的直系同源物属于拟杆菌、变形菌和厚壁菌门类。
3 CRISPR/Cas9精确识别的分子机制
3.1 可插入验证模块(pluggable authentication
modules,PAM)
PAM是Cas9系统特异识别的一个重要特征,位于靶DNA序列的3'侧翼区,在Cas9靶向结合和切割中发挥关键作用。此外,外源DNA中靶位点附近的PAM存在和宿主基因组中PAM的缺乏能使Cas9准确区分自身DNA和必须被降解的非自身DNA,尽管这两者之间可能几乎相同[37,38]。酿脓链球菌cas9的生化和结构特征表明PAM序列还参与了触发cas9与目标结合和切割构象之间的转换[32,33],表明PAM也是激活Cas9酶活性所必需的,并且决定CRISPR/Cas9系统切割发生的位点。单分子成像也证实Cas9-crRNA-tracrRNA复合体在整个基因组内首先与PAM序列结合,而后引发DNA双链的分离[38]。PAM位点和目标配体的结合可以激活HNH和Ruv C结构域从而引发Cas9的核酸酶活性,在Cas9-sgRNA-DNA三元复合体的内部可以观察到HNH的结构域为这一说法提供了有力佐证[33]。
PAM序列在不同类型CRISPR系统中存在差异,对基因组编辑效率也有重要影响。对于常用的酿脓链球菌的CRISPR/Cas9系统来说,5'-NGG是它的主要PAM序列,在人类全基因组中平均每8 bp就有一个[4]。此外,酿脓链球菌Cas9还可以靶向5'-NAG和5'-NGA PAM序列,虽然效率不高,但是这远远增加了编辑的多样性[39,40]。PAM序列对于每一个同源的Cas来说都是特异的,甚至在同一物种之中也是如此。例如,嗜热链球菌CRISPR1是5'-NAAGAAW,但是嗜热链球菌CRISPR3是5'-NGGNG[23,41]。脑膜炎奈瑟球菌的PAM序列则为5'-NNNNGATT,最近已成功被应用于人类多能干细胞的基因组编辑研究[42,43]。
PAM序列的特异性也可以被修饰,嗜热链球菌Cas9的PAM相互作用域和酿脓链球菌对应区域的同源重组可以把PAM序列从5'-NGGNG改变为5'-NGG[44]。哈佛医学院和麻省总医院的研究人员Kleinstiver等[45]建立了一套工程化系统,用进化研究的方法从大量随机突变的化脓链球菌Cas9变种中找到了可以识别新PAM序列的突变组合,这些新的Cas9变种将序列识别的范围扩大了两倍,可以识别那些野生型Cas9无法修饰的人类和斑马鱼基因的位点,这大大增加了CRISPR技术在基因组中的应用。此外,还首次证实Cas9可以用定向蛋白质进化的方法改进,利用类似的方法还可以改进Cas9酶其他的特点来满足更多的需要。
3.2 影响Cas9核酸酶特异性的主要因素
基因组编辑在基因组中能产生可遗传的修饰,因此,Cas9靶向的特异性倍受关注,尤其是在临床应用和基因治疗等方面。Cas9靶标识别是RNA与DNA的结合,其特异性由碱基互补配对决定,所以Cas9的脱靶效应相对比较容易进行实验处理分析和系统评价。近年来,多个研究小组利用错配gRNA文库、体外筛选和基因组测序广泛地鉴别了化脓链球菌Cas9的特异性,证实Cas9以一种对错配数量、位置和分布敏感的方式可以容忍sgRNA中的一些错配[39,46,47]。先前研究也同样表明Cas9的特异性是由PAM近端引导序列的前8-12个碱基核心序列决定的,其余远离PAM处12-8 bp碱基的错配对靶位点的识别影响不明显[4,29],而要想在一个基因组中产生特异性位点至少需要16个碱基的配对才可实现特异。同时也有文献报道Cas9也可以识别NAG或NNG PAM附近的序列并进行脱靶裂解,证明了在脱靶分析中也应考虑5'-NNG和5'-NAG PAM序列[31]。这些研究直接说明CRISPR/Cas9存在脱靶效应,引起基因组非靶向位点的突变,这样会造成研究结果的不确定性以及研究工作的大量增加,这一问题可能会限制Cas9的应用。而有趣的是,Cas9系统要求gRNA和靶DNA高度同源以便进行切割,但当sgRNA和靶DNA只有以小段序列互补时仍能保持短暂的结合,这表明Cas9虽然有许多潜在的脱靶位点,但是只能切割它们的一小部分[48]。因此,即使Cas9存在脱靶效应,但仍然值得开发应用于DNA结合或DNA切割。
酶的浓度也是决定Cas9脱靶发生突变的重要因素之一,因为在通常情况下靶位点上Cas9能承受5个以内的碱基错配[46],而当酶浓度较高时Cas9可承受更多的错配,这会导致脱靶位点具有更高的活性。所以,降低Cas9的浓度会显著提高靶点与脱靶位点的比率,但这是以降低靶点的切割效率为代价的[39,49]。此外,Cas9表达的持续时间可能也是调节脱靶位点活性的另一因素,但还有待深入研究。
脱靶位点一般通过搜寻与目的基因座高度同源的基因组序列来计算决定,而全基因组测序或其他全基因组范围标记双链断裂的DNA的方法进一步分析脱靶位点情况。无偏的全基因组鉴定分析已成功用于鉴定ZFNs脱靶位点的突变,这很容易被应用到Cas9核酸酶活性[50]。来自韩国首尔大学研究人员Kim等[51]开发出一种强大、敏感、无偏见和具有成本效益的方法——Digenome-seq,利用基因组测序查找CRISPR-Cas9可能突变产生的打靶和脱靶序列。这样的数据,或许结合sgRNA和靶DNA杂合链的动力学特征后,有可能为评估和预测Cas9脱靶位点活性提供了一个衡量标准。此外,耶鲁大学医学院的Moreno-Mateos等[52]分析了影响体内sgRNA稳定性、活性和装载的分子特征,确定了影响Cas9活性的决定因素,并为体内基因组打靶的高效sgRNA设计提供了一个框架,构建了一种预测性的sgRNA评分算法——CRISPRscan,可有效地捕捉到影响CRISPR/Cas9体内活性的序列特征。
3.3 提高Cas9靶位点识别精确度的策略
为了提高靶位点DNA双链断裂的特异性,可以通过类似二聚ZFNs和TALENs的双切口方法来增加在目的基因中可以特异识别的碱基数目。使用一对gRNA和一个酿脓链球菌Cas9 HNH+/RuvC-切口酶突变体(D10A),适当的隔开协同的切口可以模拟DNA双链断裂高效介导插入缺失的形成,能有效减少意外的脱靶基因修饰[47,53]。因为它分别在相对的两条DNA链上产生切口,从而形成功能性的双链断裂,这种设计能最大限度降低脱靶效应,同时保持高效而又特异的基因修饰,这种策略可以将有关野生型Cas9的特异性提高50-1 500倍[47,53]。
除了双切口策略,截短的sgRNA可以通过减少2-3个核苷酸来显著增加酿脓链球菌Cas9靶向的特异性,可能是由更高的错配敏感度造成的[54]。这些被截短的sgRNA可以和多切口策略结合,进一步降低脱靶突变。另外,未来对Cas9的结构功能分析和通过合理设计或定向进化的蛋白质工程可能会进一步提高Cas9的特异性。
NHEJ和同源介导修复(homology-directed,HDR)是几乎所有的细胞和生物体都利用的两种DNA修复机制,相比HDR细胞往往更依赖NHEJ,然而,在使用CRISPR/Cas9时HDR则是首选的、更精确的机制。但以往的应用中CRISPR/Cas介导的HDR效率远远低于20%,因此,不利于通过生成足够数量的多基因精确突变转基因小鼠来构建人类疾病模型。为了提高HDR的活性,Maruyama等[55]将抗癌药物Scr7添加到细胞(经过遗传编辑的受精卵)中,Scr7可与NHEJ信号通路中的一个关键酶Ligase IV结合,从而阻止经由NHEJ的DNA修复,因此,细胞内的DNA修复主要由更为精确的HDR机制来接管,这一过程中CRISPR/Cas的效率显著提高了19倍。这种基于药物的方法真正地改进科学家构建转基因小鼠的方式,使之携带上想研究的特异突变,生成更复杂突变的变异小鼠。
最近,来自哈佛大学、麻省总医院的研究人员Davis等[56]提出对靶向位点进行修饰可以直接调控及降低基因组编辑蛋白的活性,这有可能成为提高它们特异性的一种潜在方法。他们通过将4-羟基他莫昔芬(hydroxytamoxifen)反应性的内含肽插入到Cas9的特异位点,开发出了可被一种细胞渗透性小分子激活的Cas9核酸酶。证实在人类细胞中,条件性激活的Cas9将改造靶基因组位点的特异性提高了25倍。相信通过这些及其他新的策略来不断提高Cas9的特异性,Cas9未来在临床及基因治疗等方面的应用将是令人期待的。
4 CRISPR/Cas9技术在基因组编辑中的应用
3类CRISPR系统中TypeⅡ型系统的组成相对简单,除crRNA和tracrRNA外,只有Cas9一个蛋白,其具体作用机制也比其他类型CRISPR系统研究的更为清楚,因此对动植物基因组的编辑过程中都是采用该类型。2013 年初,发表在同一期《Science》研究论文报道了来自于麻省理工学院的Zhang等和哈佛大学的Mali等利用CRISPR/Cas9系统在小鼠和人类细胞中首次实现了基因组的定点编辑,并且获得较高的打靶效率[4,5]。随后,在世界范围内掀起了CRISPR/Cas9系统用于基因编辑研究的热潮,在短短的几年里,CRISPR/Cas9系统被广泛的应用到许多物种和各类细胞中,成功地对目的基因进行定向缺失、插入、活化或抑制等遗传改造操作,其中包括人类、细菌、斑马鱼、酵母、小鼠、果蝇、农作物、家畜和猴子[8,57,58]。CRISPR/Cas9大大增加了遗传上易处理的模式生物的数目。例如,南京大学模式动物研究所等的研究人员成功地利用CRISPR/Cas9系统对孪生的食蟹猴进行了精确的基因修饰[44]。这是人类首次利用基因编辑技术在灵长类动物中获得成功,使得对灵长类动物的研究前景变得光明,这一重要发现于将来某一天能够帮助构建出与多种突变相关的其他人类疾病模型。
4.1 细胞和动物模型的快速制备
Cas9介导的基因组编辑可以加快转基因模型的产生,扩展超越了传统生物学研究,更容易得到可遗传模式生物。例如,通过重现患者群体中的基因突变,基于CRISPR/Cas9的编辑可以用来快速建立特异的遗传突变模型,而不是依赖疾病模型中表型模拟一个特殊的病症[44,59]。这可以应用于开发新的转基因动物模型,来设计带有引入或修正特异基因突变的ES和iPS疾病模型,或者在体内和体外进行基因修正[60]。
Cas9能够通过质粒和sgRNA一起瞬时转染很容易导入到靶细胞,产生需要的细胞模型。此外,Cas9的多种功能为研究人类常见疾病提供了一种新的方法,如糖尿病、心脏病、精神分裂症和自闭症等。例如,大规模的全基因组关联研究(GWAS)在鉴别与疾病风险密切相关的单倍型上发挥了重要作用,但通常很难确定在紧密连锁不平衡中哪几个遗传变异和这个区域的单倍体中哪些基因控制表型。利用CRISPR/Cas系统,人们可以通过编辑干细胞并使其分化成我们想要的细胞类型的方法来研究每个突变体的结果,或者在等位基因的背景下测试单个处理的效果,从而精确确定哪些基因控制表型[61]。
Cas9蛋白和转录的sgRNA还可以直接被注射到小鼠的受精卵中来获得可遗传的基因修饰,从而获得转基因动物模型,如一个或多个等位基因修饰的啮齿动物和猴子模型[44,59,62,63]。通过传统的胚胎干细胞来获得转基因品系,得到小鼠和大鼠突变体的时间将由一 年减少到短短几周。这样的改进使得在啮齿目动物模型中进行大规模的体内诱变研究变得更经济,并且能与高特异性编辑结合从而避免脱靶诱变的混淆[53,54]。此外,Cas9能够利用对体细胞的直接修饰从而无需胚胎操作即可实现基因治疗。
然而,通过受精卵注射CRISPR试剂产生的转基因动物模型的一个突出的挑战是遗传嵌合体,部分原因是由于核酸酶诱导的诱变速率缓慢。迄今为止,有关转基因动物模型的研究都普遍依赖于向受精卵(胚胎受精的单细胞阶段)中注射Cas9。由于转录和翻译活动在小鼠的受精卵中都受到抑制,Cas9 mRNA转换成酶的形式很可能被延迟到第一次细胞分裂之后[64]。而NHEJ介导的修复作用是引入随机长度的插入或缺失DNA片段,这个翻译延迟很可能是CRISPR修饰小鼠形成遗传嵌合体的主要原因。为了克服这个局限,Cas9蛋白和sgRNA可以被直接注射到单细胞受精卵中,通过NHEJ介导的DNA修复过程可能会减少一些我们不希望有的遗传嵌合体,因为向Cas9识别位点引入插入或缺失突变将不得不与受精卵的分裂速度竞争。
4.2 功能基因组学筛选
随着对CRISPR/Cas9工具的进一步开发,该系统已用于全基因组功能筛选,以极高的灵敏度和精密度帮助研究人员发掘新的基因,并精确鉴定哪种或哪类基因在期望的表型汇总中扮演重要角色。例如,使用具有成千上万个gRNA(可靶定人类或其他物种基因组中所有功能基因)的CRISPR/cas9系统,可让研究人员在基因组规模上进行功能获得或功能缺失突变筛选,再通过快速扫描基因组中所有的基因,包括来自各种各样试验的新候选基因。北京大学的研究人员构建了一套基于新型CRISPR/Cas9 sgRNA文库的高效遗传筛选技术[65],这项技术主要包括预先建立稳定表达Cas9的细胞系并确定sgRNA能够在该细胞系实现基因修饰、建立sgRNA慢病毒文库以及细胞文库、功能性筛选和富集、PCR扩增和深度测序分析、数据分析及候选基因的验证等几个步骤。通过该技术,研究人员成功地鉴别出了对于炭疽和白喉毒素毒性至关重要的宿主基因,并在随后的细胞实验中对这些候选基因进行了进一步的功能验证。该研究发现了与病菌侵染宿主蛋白及通路有关的基因,这将为对抗病菌提供新型药物靶点,而且更重要的是,这一强大的高通量基因筛选技术的建立可以广泛应用于生物学问题的研究,惠及众多生物医学相关领域。此外,其他一些相关研究小组也分别报道了与之相类似的技术路线,通过构建哺乳细胞CRISPR/Cas9敲除文库实现高通量功能性基因的筛选[66-68]。
sgRNA文库未来还可能应用于基因组非编码元件的编辑,如基因调节区的系统靶向可促进远端增强子、一般启动子结构以及对转录水平有影响的任何额外调节元件的发现[31]。此外,还可以应用于研究大量的无具体特征的基因组区域,这些序列研究或大规模的全基因组关联研究(GWAS)中都有涉及。
4.3 转录调控
Cas9-sgRNA作为一种可与靶序列特异性识别并切割的技术,其应用不仅仅局限于对靶序列的精确编辑,还可拓展到基因转录调控方面。通过点突变使Cas9蛋白的RuvC和HNH核酶结构域活性全部丧失获得dCas9(Catalytically dead Cas9)的能力,但dCas9仍保留了与靶序列特异性结合的能力,这样可将dCas9-sgRNA作为与DNA特异性识别的平台,同样具有较大的应用价值。dCas9-sgRNA与DNA转录元件结合可能会对RNA聚合酶造成空间位阻,从而阻断该位点上的基因转录,起到转录抑制的效果。例如,在dCas9蛋白的C末端融合转录抑制结构域(krüppel associated box,KRAB),获得的dCas9-KRAB-sgRNA具有更强的转录抑制效果,将可以替代脱靶效应明显的siRNA技术,称之为CRISPR干扰或CRISPRi[69,70]。dCas9蛋白的C末端还可融合转录激活结构域疹病毒转录激活子VP16或NF-κB p65亚基活性域AD(p65AD),则可构建具有转录激活活性的CRISPR-on系统,用于激活特定靶基因的转录[70-72]。通过以上这两个方面的改造可以使Cas9-sgRNA 成为对基因表达的调控工具。但这些系统都是组成型的,无法打开和关闭转录本,2015年,3个研究小组各自均开发出了基于CRISPR-Cas9的转录激活系统,可以用光进行控制,很好地解决了这一难题[73-75]。该系统包括一对融合蛋白,一个蛋白把dCas9结合到一个称为CIb1的蛋白;另外一个蛋白将一个转录激活结构域结合到隐花色素2(CRY2),用蓝光照亮表达这两个蛋白质的细胞和gRNA,可使两个蛋白质配对,将转录激活结构域拴在DNA上,并激活转录。该设计为以前这种构成性的合成转录因子引入了一种调控机制,该方法可通过光激活靶向的、用户界定的基因,可有助于众多生物医学的应用,如利用其在体内外精确控制细胞的功能。
4.4 细胞内基因组的活体成像
细胞内功能和结构元件的空间组织对基因组的功能性输出能够动态的增强或抑制作用,然而基因组被修饰的方式以及在体内结构组织调节功能的方式仍然不清楚。开发用于能直接监控活体生物体内的细胞活动和基因行为至关重要,目前现有技术主要有染色质构象捕获(3C)和使用高分辨率显微镜的荧光原位杂交(fluorescence in situ hybridization,FISH)分析,但这些技术很难探索基因组和基因表达随时间而发生的空间组织变化,动态信息的获取,对高阶基因调控的全面了解是必需的。因此,迫切需要开发新的技术来分析基因转录和空间组织的动力学。目前,基于CRISPR/Cas9的特定DNA位点的荧光标记Cas9标签替代了DNA-FISH技术发展成了一种功能强大的活细胞成像技术[76],这将用于研究复杂的染色体结构和核组织。Ochiai等[77]建立了一种被称为“Real-time Observation of Localization and Expression(ROLEX)”系统的新的实时成像方法,该系统可同时测量内源基因的转录活性和核定位,具有很高的特异性,且不影响靶基因的表达水平。ROLEX系统可用于研究通过远程基因组相互作用的高阶基因调控动力学,揭示活细胞中基因转录和细胞核动态之间的关系,并评估细胞的基因组结构变异,是否会引起基因表达中的细胞间异质性。无疑ROLEX将会成为一种强大的工具,不仅用于细胞生物学,也可应用于生物医学研究和临床疗法等领域。
4.5 遗传性疾病的治疗
CRISPR/Cas9在短短几年的发展中,在从基础研究到农业领域以及生物医学方面都展现出强大的应用潜力,但Cas9作为一种基因治疗技术,用于彻底根治遗传性疾病是未来发展方向。利用CRISPR/ Cas9技术通过体细胞基因组编辑,纠正致病突变,可以直接治疗有害的遗传疾病。例如,对于由功能缺失突变引起的单基因隐性遗传病(如纤维囊泡症、镰状细胞性贫血及杜氏肌营养不良症等),Cas9可能用于修正致病突变。Long等[78]采用CRISPR/Cas9技术成功阻止了杜氏肌营养不良小鼠模型中的肌肉退化,研究人员移除了携带突变dystrophin的小鼠的胚胎,并在胚胎中注射了靶向纠正dystrophin突变的CRISPR编辑元件,然后将胚胎返回到野生型雌鼠体内,这样子代小鼠具有正常的dystrophin和骨骼肌功能,而且虽然只有17%的细胞得到了纠正,但也能令其骨骼肌恢复功能。尽管这种技术目前未应用于人体,但是这一概念为未来治疗肌营养不良症带来了希望。而中国科学家首次实现了CRISPR/Cas9技术用于编辑人类胚胎基因组,改造了导致一种潜在致命血液疾病——β-地中海贫血的基因(HBB基因),但编辑成功率较低[79],这让人们看到通过胚胎基因编辑治疗遗传性疾病的光明未来。
有些单基因疾病是由于基因组序列的重复引起的,对于这些疾病,用Cas9可以删除这些重复元件。例如,三核苷酸重复疾病可以通过同时用两个DSBs来切除重复区域进行处理达到治疗目的[80]。此策略的成功有可能治疗更复杂的遗传性疾病如弗里德赖希共济失调症(在致病基因的非编码区存在较多三核苷酸重复)[81]。
此外,为了修复潜在的遗传性疾病,Cas9介导的基因组编辑可在体细胞或组织中引入一种保护性突变来对抗非基因或复杂的疾病。例如,NHEJ介导的淋巴细胞中CCR5受体的失活可能对防止艾滋病感染是一种不错的策略[82],而缺失PCSK9或血管生成素可能有抵抗高胆固醇血症或高血脂症的疗效[83,84]。虽然这些目标也可通过siRNA介导的蛋白质抑制达到,但NHEJ介导的基因失活无需持续治疗即可获得永久疗效。如同所有的基因疗法,用低风险率的方法建立各自的治疗方法非常重要。
尽管基因组编辑为基础的遗传性疾病的彻底根治带来希望,许多研究团体正努力使CRISPR/Cas9疗法成为现实,但众多的挑战依然摆在面前。成功的临床治疗取决于向特定的疾病组织靶标中恰当高效的注入CRISPR/Cas9系统,而为了获得高效的治疗效果和同时治疗多种遗传病,高效的同源重组需要高度重视。即使永久的遗传修饰对于单克隆抗体或siRNA治疗来说具有优势,但其需要对分子治疗进行反复实验,其长期影响尚不清楚。此外,研究人员需进一步测试用于临床治疗的Cas9,用多种多样的临床模型来证明Cas9的安全性和生理效应是十分重要的。
4.6 病毒感染性疾病的治疗
病毒通过特异的受体侵染细胞,在细胞内发生复制、转录、翻译等过程完成其生活周期,病毒感染疾病是危害人类健康的主要威胁之一,与人类密切相关的有呼吸道病毒、疱疹病毒、肝炎病毒和艾滋病病毒等。目前治疗病毒感染性疾病主要依赖疫苗防御和药物治疗,但很多还没有完全有效或彻底的治疗方法,如艾滋病等。CRISPR/Cas9随着基因定点编辑技术的成熟与完善,使得抗病毒治疗逐渐成为可能。美国坦普尔大学Hu等[85]构建了靶向HIV-1启动子的gRNA-Cas9质粒,转染整合到有HIV感染的小胶质细胞、巨噬细胞以及T淋巴细胞,均成功地根除了HIV基因组,表明该技术具有彻底清除潜伏的HIV基因组的可能性。尽管CRISPR/ Cas9可以对培养的感染细胞中的HIV进行编辑,但这种技术的精度仍然不足以应用于临床,因其往往切入基因组的随机区域,易于产生有害的、非靶目标效应,还有待进一步提高精准度。此外,相似的研究也被应用于疱疹病毒[86]、乙型肝炎病毒[87]、丙型肝炎病毒[88]及人乳头瘤病毒[89]等基因组的编辑,并已达到彻底清除病毒的目的,无疑给这些病毒感染性疾病的彻底治疗带来希望。
5 总结
短短的几年,由在细菌和古细菌等原核生物中发现的遗传物质靶向编辑系统演变而来的CRISPR/ Cas9技术取得了令人欣喜的发展,使得研究人员能快速地对遗传物质进行随心所欲地操作或修饰,与成熟的ZFN和TALEN等基因靶向编辑系统相比,CRISPR/Cas9系统具有得天独厚的优越性,如构建简单、方便、快捷,安全性高、毒性小等。相信基于Cas9的基因编辑工具极有可能是未来进行精确和高效的基因修饰的解决办法,在临床治疗、基础理论研究和农牧渔业等领域必将发挥巨大的作用,并且将会对分子生物学研究和基因治疗领域产生深远的影响。
[1]Smith J, Grizot S, Arnould S, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences[J]. Nucleic Acids Res, 2006, 34(22):e149.
[2]Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases[J]. Nature, 2005, 435(7042):646-651.
[3]Boch J, Scholze H, Schornack S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors[J]. Science, 2009, 326(5959):1509-1512.
[4]Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013, 339(6121):819-823.
[5]Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9[J]. Science, 2013, 339(6121):823-826.
[6]Maeder ML, Thibodeau-Beganny S, Osiak A, et al. Rapid “opensource” engineering of customized zinc-finger nucleases for highly efficient gene modification[J]. Mol Cell, 2008, 2:294-301.
[7]Gonzalez B, Schwimmer LJ, Fuller RP, et al. Modular system for the construction of zinc-finger libraries and proteins[J]. Nat Protoc, 2010, 5(4):791-810.
[8]Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes[J]. Nat Biotechnol, 2014, 4:347-355.
[9]Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system[J]. Proc Natl Acad Sci USA, 2015, 11:3570-3575.
[10]Ishino Y, Shinagawa H, Makino K, et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product[J]. J Bacteriol, 1987, 169(12):5429-5433.
[11]Mojica FJ, Diez-Villasenor C, Soria E, et al. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria[J]. Mol Microbiol, 2000, 36(1):244-246.
[12]Jansen R, Embden JD, Gaastra W, et al. Identification of genes that are associated with DNA repeats in prokaryotes[J]. Mol Microbiol, 2002, 43(6):1565-1575.
[13]Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes[J]. Science, 2007, 315(5819):1709-1712.
[14]Haft DH, Selengut J, Mongodin EF, et al. A guild of 45 CRISPR-associated(Cas)protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genome s[J]. PLoS Comput Biol, 2005, 1(6):e60.
[15]Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR-Cas systems[J]. Nat Rev Microbiol, 2011, 9(6) :467-477.
[16]Chylinski K, Makarova KS, Charpentier E, et al. Classification and evolution of type II CRISPR-Cas systems[J]. Nucleic Acids Res, 2014, 42(10):6091- 6105.
[17]Bolotin A, Quinquis B, Sorokin A, et al. Clustered regularly interspaced short palindrome repeats(CRISPRs)have spacers of extrachromosomal orig in[J]. Microbiology, 2005, 151(Pt 8):2551-2561.
[18]Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, et al. Intervening seque nces of regularly spaced prokaryotic repeats derive from foreign genetic elements[J]. J Mol Evol, 2005, 2:174-182.
[1 9]Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies[J]. Microbiology, 2005, 151(Pt 3):653-663.
[20]Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes[J]. Science, 20 08, 321(5891):960-964.
[21]Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by tar geting DNA[J]. Science, 2008, 322(5909):1843-1845.
[22]Hale CR, Zhao P, Olson S, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex[J]. Cell, 2009, 139(5):945-95 6.
[23]Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus[J]. J Bacteriol, 2008, 190(4):1390-1400.
[24]Horvath P, Coute-Monvoisin AC, Romero DA, et al. Comparative analysis of CRISPR loci in lactic acid bacteria genomes[J]. Int J Food Microbiol, 2009, 131(1):62-70.
[25]Garneau JE, Dupuis ME, Villion M, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plas mid DNA[J]. Nature, 2010, 468(7320):67-71.
[26]Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III[J]. Nature, 2011, 471(7340):602-607.
[27]Sapranauskas R, Gasiunas G, Fremaux C, et al. The Streptococcus thermophilus CRISPR/Cas system provides imm unity in Escherichia coli[J]. Nucleic Acids Res, 2011. 39(21):9275-9282.
[28]Gasiunas G, Barrangou R, Horvath P, et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptiv e immunity in bacteria[J]. Proc Natl Acad Sci USA, 2012, 109(39):E2579-2586.
[29]Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J]. Science, 2012, 337(6096):816-821.
[30]Doudna JA, Charpentier E. Genome editing. The new frontier o f genome engineering with CRISPR-Cas9[J]. Science, 2014, 346(6213):1258096.
[31]Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering[J]. Cell, 2014, 157(6) :1262-1278.
[32]Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation[J]. Science, 2014, 343(6176):1247997.
[33]Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA[J]. Cell, 2014, 156(5):935-949.
[34]Jiang F, Zhou K, Ma L, et al. A Cas9-guide RNA complex preorganized for target DNA recognition[J]. Science, 2015, 348( 6242):1477-1481.
[35]Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems[J]. RNA Biol, 2013, 10(5):726-737.
[36]Fonfara I, Le Rhun A, Chylinski K, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among ort hologous type II CRISPR-Cas systems[J]. Nucleic Acids Res, 2014, 42(4):2577-2590.
[37]Shah SA, Erdmann S, Mojica FJ, et al. Prot ospacer recognition motifs:mixed identities and functional diversity[J]. RNA Biol, 2013, 10(5):891-899.
[38]Sternberg SH, Redding S, Jinek M, et al. DNA interr ogation by the CRISPR RNA-guided endonuclease Cas9[J]. Nature, 2014, 507(7490):62-67.
[39]Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guid ed Cas9 nucleases[J]. Nat Biotechnol, 2013, 31(9):827-832.
[40]Jiang W, Bikard D, Cox D, et al. RNA-guided editing of bacterialgenomes using CRISPR-Cas syst ems[J]. Nat Biotechnol, 2013, . 31(3):233-239.
[41]Horvath P, Romero DA, Coute-Monvoisin AC, et al. Diversity, activity, and evolution of CRISPR loci in Strept ococcus thermophilus[J]. J Bacteriol, 2008, 190(4):1401-1412.
[42]Zhang Y, Heidrich N, Ampattu BJ, et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis[J]. Mol Cell, 2013, 50(4):488-503.
[43]Hou Z, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis[J]. Proc Natl Acad Sci USA, 2013, 110(39):15644-15649.
[44]Niu Y, Shen B, Cui Y, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in onecell embryos[J]. Cell, 2014, 156(4):836-843.
[45]Kleinstiver BP, Prew MS, Tsai SQ, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities[J]. Nature, 2015, 523(7561):481-485.
[46]Fu Y, Foden JA, Khayter C, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells[J]. Nat Biotechnol, 2013, 31(9):822-826.
[47]Mali P, Aach J, Stranges PB, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering[J]. Nat Biotechnol, 2013, 9:833-838.
[48]Wu X, Scott DA, Kriz AJ, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells[J]. Nat Biotechnol, 2014, 32(7):670-676.
[49]Pattanayak V, Lin S, Guilinger JP, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity[J]. Nat Biotechnol, 2013, 9:839-843.
[50]Gabriel R, Lombardo A, Arens A, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity[J]. Nat Biotechnol, 2011, 29(9):816-823.
[51]Kim D, Bae S, Park J, et al. Digenome-seq:genome-wide profiling of CRISPR-Cas9 off-target effects in human cells[J]. Nat Methods, 2015, 12(3):237-243.
[52]Moreno-Mateos MA, Vejnar CE, Beaudoin JD, et al. CRISPRscan:designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo[J]. Nat Methods, 2015, 12(10):982-988.
[53]Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity[J]. Cell, 2013, 154(6):1380-1389.
[54]Fu Y, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs[J]. Nat Biotechnol, 2014, 32(3):279-284.
[55]Maruyama T, Dougan SK, Truttmann MC, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining[J]. Nat Biotechnol, 2015, 33(5):538-542.
[56]Davis KM, Pattanayak V, Thompson DB, et al. Small moleculetriggered Cas9 protein with improved genome-editing specificity[J]. Nat Chem Biol, 2015, 11(5):316-318.
[57]Peng J, Zhou Y, Zhu S, et al. High-throughput screens in mammalian cells using the CRISPR-Cas9 system[J]. FEBS J, 2015, 282(1):2089-2096.
[58]Wiles MV, Qin W, Cheng AW, et al. CRISPR-Cas9-mediated genome editing and guide RNA design[J]. Mamm Genome, 2015, 26(9-10):501-510.
[59]Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell, 2013, 153(4):910-918.
[60]Smith C, Abalde-Atristain L, He C, et al. Efficient and allelespecific genome editing of disease loci in human iPSCs[J]. Mol Ther, 2015, 23(3):570-577.
[61]Kearns NA, Genga RM, Enuameh MS, et al. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells[J]. Development, 2014, 1:219-223.
[62]Li W, Teng F, Li T, et al. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems[J]. Nat Biotechnol, 2013, 31(8):684-686.
[63]Yang H, Wang H, Shivalila CS, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering[J]. Cell, 2013, 154(6):1370-1379.
[64]Oh B, Hwang S, McLaughlin J, et al. Timely translation during the mouse oocyte-to-embryo transition[J]. Development, 2000, 127(17):3795-3803.
[65]Zhou Y, Zhu S, Cai C, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells[J]. Nature, 2014, 509(7501):487-491.
[66]Koike-Yusa H, Li Y, Tan EP, et al. Genome-wide recessive geneticscreening in mammalian cells with a lentiviral CRISPR-guide RNA library[J]. Nat Biotechnol, 2014, 32(3):267-273.
[67]Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPRCas9 knockout screening in human cells[J]. Science, 2014, 343(6166):84-87.
[68]Wang T, Wei JJ, Sabatini DM, et al. Genetic screens in human cells using the CRISPR-Cas9 system[J]. Science, 2014, 343(6166):80-84.
[69]Long L, Guo H, Yao D, et al. Regulation of transcriptionally active genes via the catalytically inactive Cas9 in C. elegans and D. rerio[J]. Cell Res, 2015, 25(5):638-641.
[70]Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes[J]. Cell, 2013, 154(2):442-451.
[71]Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology[J]. Nat Methods, 2013, 10(10):957-963.
[72]Perez-Pinera P, Kocak DD, Vockley CM, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors[J]. Nat Methods, 2013, 10(10):973-976.
[73]Nihongaki Y, Yamamoto S, Kawano F, et al. CRISPR-Cas9-based photoactivatable transcription system[J]. Chem Biol, 2015, 22(2):169-174.
[74]Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation[J]. Nat Chem Biol, 2015, 11(3):198-200.
[75]Zetsche B, Volz SE, Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation[J]. Nat Biotechnol, 2015, 33(2):139-142.
[76]Chen B, Gilbert LA, Cimini BA, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system[J]. Cell, 2013, 155(7):1479-1491.
[77]Ochiai H, Sugawara T, Yamamoto T. Simultaneous live imaging of the transcription and nuclear position of specific genes[J]. Nucleic Acids Res, 2015, 43(19):e127.
[78]Long C, McAnally JR, Shelton JM, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA[J]. Science, 2014, 345(6201):1184-1188.
[79]Liang P, Xu Y, Zhang X, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes[J]. Protein Cell, 2015, 6(5):363-372.
[80]Richard GF. Shortening trinucleotide repeats using highly specific endonucleases:a possible approach to gene therapy?[J]. Trends Genet, 2015, 31(4):177-186.
[81]Vannocci T, Faggianelli N, Zaccagnino S, et al. A new cellular model to follow Friedreich’s ataxia development in a timeresolved way[J]. Dis Model Mech, 2015, 8(7):711-719.
[82]Lombardo A, Genovese P, Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrasedefective lentiviral vector delivery[J]. Nat Biotechnol, 2007, 25(11):1298-1306.
[83]Cohen J, Pertsemlidis A, Kotowski IK, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9[J]. Nat Genet, 2005, 37(2):161-165.
[84]Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia[J]. N Engl J Med, 2010, 363(23):2220-2227.
[85]Hu W, Kaminski R, Yang F, et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection[J]. Proc Natl Acad Sci USA, 2014, 111(31):11461-11466.
[86]Bi Y, Sun L, Gao D, et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases[J]. PLoS Pathog, 2014, 10(5):e1004090.
[87]Lin SR, Yang HC, Kuo YT, et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo[J]. Mol Ther Nucleic Acids, 2014, 3:e186.
[88]Price AA, Sampson TR, Ratner HK, et al. Cas9-mediated targeting of viral RNA in eukaryotic cells[J]. Proc Natl Acad Sci USA, 2015, 112(19):6164-6169.
[89]Zhen S, Hua L, Takahashi Y, et al. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9[J]. Biochem Biophys Res Commun, 2014, 450(4):1422-1426.
(责任编辑 狄艳红)
The Development of CRISPR/Cas9 Technique and Its Applications in Genome Editing
ZHANG Kai-li LI Rui HU Tong-tong XU Yong-jie
(College of Life Sciences,Xinyang Normal University,Xinyang 464000)
CRISPR/Cas9 is one of novel genome editing technologies developed from the unique prokaryotic acquired immune system in bacteria and archaea,and it can be guided to specific locations within complex genomes by a short RNA,and thus cleavage of the target can be recognized. Using this technology,DNA sequences within the endogenous genome and their expressed products are now easily edited or regulated in virtually any organism of choice. The technology has become the most popular genome editing tool,promoting the development of basic biology,biotechnology and medicine. This paper introduced the research progress on CRISPR/Cas9 from the aspects of development history,structure and function,and the mechanism of precise recognition,also reviewed the application of this new technology in genome editing in detailed,aiming at providing a reference for related researchers.
engineered endonuclease;Cas9;PAM;CRISPR/Cas;genome editing
10.13560/j.cnki.biotech.bull.1985.2016.05.007
2015-08-08
国家自然科学基金项目(U1204326),国家大学生创新训练项目(20150477021)
张凯丽,女,硕士研究生,研究方向:动物分子遗传;E-mail:kaili_scc@sina.com
徐永杰,男,博士,副教授,究方向:动物分子遗传;E-mail:yongjx81@126.com
猜你喜欢
今日农业(2021年11期)2021-08-13
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
广州大学学报(自然科学版)(2019年1期)2019-05-07
中国病理生理杂志(2017年2期)2017-01-17
天津科技大学学报(2016年1期)2016-02-28
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
现代检验医学杂志(2015年2期)2015-02-06
遗传(2014年3期)2014-02-28
