
β-环糊精/硅基杂化手性固定相的制备及其手性拆分性能
2016-06-22 00:47王利涛董树清张志欣王杨军张晓莉张鹏云
色谱 2016年1期
王利涛, 董树清, 张志欣, 王杨军, 张晓莉, 张 霞, 张鹏云,3, 赵 亮*
(1. 中国科学院西北特色植物资源化学重点实验室, 甘肃省天然药物重点实验室, 中国科学院兰州化学物理研究所, 甘肃 兰州 730000; 2. 西北师范大学, 甘肃 兰州 730000; 3. 甘肃省化工研究院, 甘肃 兰州 730000)
β-环糊精/硅基杂化手性固定相的制备及其手性拆分性能
王利涛1,董树清1,张志欣1,王杨军2,张晓莉1,张霞1,张鹏云2,3,赵亮1*
(1. 中国科学院西北特色植物资源化学重点实验室, 甘肃省天然药物重点实验室, 中国科学院兰州化学物理研究所, 甘肃 兰州 730000; 2. 西北师范大学, 甘肃 兰州 730000; 3. 甘肃省化工研究院, 甘肃 兰州 730000)
摘要:发展了一种制备β-环糊精/硅基杂化手性固定相的简单方法。首先合成了β-环糊精硅基衍生物,然后在碱性条件下通过硅烷化试剂和β-环糊精硅基衍生物之间的聚合反应,制备了β-环糊精衍生物共价负载于孔道表面的球形β-环糊精/硅基杂化材料,去除模板剂,即可得到直接用于高效液相色谱填料的β-环糊精/硅基杂化手性固定相。制备的杂化固定相材料具有球形规则、单分散性好、比表面积高、机械性能好、化学稳定性高和制备过程简单等特点,结合了β-环糊精的手性识别功能与有机-无机杂化材料的优异性能。在反相色谱条件下的手性拆分结果表明杂化手性固定相具有较高的手性识别能力。本文所发展的方法为新型手性固定相的制备提供了一种新的思路。
关键词:高效液相色谱;手性固定相;β-环糊精;有机-无机杂化;手性拆分
手性固定相是高效液相色谱手性分离的核心,在对映体的分离过程中起关键作用。手性固定相的研发是高效液相色谱手性分离材料发展的前沿和核心领域[1,2]。环糊精键合手性固定相可以在正相和反相色谱条件下分离非常广泛的手性客体,是高效液相色谱拆分手性化合物的重要工具[3-5]。近年来,尽管环糊精键合手性固定相的制备、拆分过程以及拆分机理等方面的研究取得了巨大进展,但还存在一些问题[6-8],如环糊精的自身体积大,位阻效应强,导致环糊精键合量偏低;硅胶表面的硅羟基数量有限,仅为7~8 μmol/m2;硅胶中键合的环糊精分布不均匀,在孔口部分拥挤,容易导致部分孔堵塞,而在内孔壁的环糊精分布偏低,影响环糊精与被分析物之间的相互作用;环糊精键合手性固定相的制备过程繁琐,成本高。因此,新型高效液相色谱环糊精手性固定相的开发是当前手性拆分色谱技术中的重要研究内容。
有机-无机杂化介孔材料具有比表面积大、孔体积大、孔道尺寸可调以及特殊的有机功能基团等特性,在液相色谱固定相方面具有广泛的应用潜力[9-11]。将环糊精作为功能基团引入到有机-无机杂化材料的孔道中一直受到人们的关注。早在2001年,Mercier等[12]以较高的负载量将β-环糊精引入到硅基杂化材料的介孔孔道中,但是并没有获得球形的形貌。Degoutin等[13]也成功地将β-环糊精引入到硅基杂化材料的孔道中,虽没有得到球形的形貌,但是可以用于去除废水中的农药、染料和腐殖酸。近年来,Zou等[14]制备了β-环糊精/硅基有机-无机杂化毛细管整体柱,在反相色谱条件下成功拆分了13种对映体。这表明在孔道中负载了β-环糊精的杂化材料具有很高的手性识别能力。因此,制备具有球形形貌和粒径均匀的β-环糊精/硅基有机-无机杂化材料是一项有挑战性的工作,并且该材料作为高效液相色谱手性固定相也具有重要的应用潜力。
本课题组前期通过“一锅法”制备了β-环糊精/硅基杂化硅球,并将环糊精羟基进行了3,5-二甲基苯基衍生,制备了β-环糊精/硅基杂化多功能手性固定相[15],在正/反相色谱模式下,通过多种作用力拆分了多种手性化合物,并分离了苯胺类、羧酸类和多苯环类化合物,表现出了良好的手性识别和色谱分离性能。本文通过两步化学反应,首先合成了β-环糊精硅基衍生物,然后以十六烷基三甲基溴化铵为模板剂,以β-环糊精硅基衍生物与1,2-双(三乙氧基硅基)乙烷为混合硅源,通过有机-无机杂化技术制备了β-环糊精/硅基杂化介孔硅球作为高效液相色谱手性固定相。在反相色谱模式下,评价了β-环糊精/硅基杂化介孔硅球拆分对映体的性能,发展了一类制备过程简单的新型β-环糊精/硅基杂化手性固定相。本文将β-环糊精的手性识别功能与有机-无机杂化材料的优异性能相结合,制备新型有机-无机杂化环糊精手性固定相,实现手性药物的快速、高效分离,在手性药物拆分研究方面有重要的应用价值。
1实验部分
1.1试剂与材料
一氯三嗪-β-环糊精(分析纯,南京都莱生物技术有限公司); 1,2-双(三乙氧基硅基)乙烷(分析纯,美国Gelest公司);氨丙基三乙氧基硅烷(分析纯,武汉天目科技发展有限责任公司);十六烷基三甲基溴化铵和氢氧化钠(分析纯,中国医药集团上海化学试剂公司); 2-苯基-1-丙醇和1-苯乙醇和普萘洛尔(分析纯,美国Sigma-Aldrich公司);甲磺酸帕苏沙星和甲磺酸氧氟沙星(分析纯,百灵威试剂公司);甲醇(MeOH)和乙腈(ACN)(色谱纯,山东禹王实业有限公司);三乙胺和醋酸(分析纯,天津化学试剂二厂)。
1.2仪器
高效液相色谱系统(包括Waters公司515泵、2487检测器,Rheodyne公司7725i手动进样器,Millennium32软件); Vairo元素分析仪(德国Elementar公司); IFS120HR傅里叶变换红外光谱仪(德国Bruker公司); 95551U装柱机(美国Alltech公司);不锈钢色谱柱管(150 mm×4.6 mm,天津三维色谱仪器配件厂); ASAP 2010快速比表面积/孔隙分析仪(美国Micromeritics公司); JSM-5601LV扫描电子显微镜(日本电子株式会社); TF20场发射透射电子显微镜(美国FEI公司); Milli-Q Integral超纯水处理系统(美国Millipore公司)。
1.3β-环糊精/硅基杂化手性固定相的制备
β-环糊精/硅基杂化球的具体制备过程如图1所示。将一氯三嗪-β-环糊精1.87 g和氨丙基三乙氧基硅烷0.78 mL置于20 mL水中,再加入0.1 g NaHCO3,在40 ℃水浴中磁力搅拌30 min。加入80 mL乙醇,使沉淀析出。抽滤,乙醇洗涤,真空干燥,即可得到β-环糊精硅基衍生物。
取β-环糊精硅基衍生物1.8 g加入到含有22 mL水、7 mL乙醇、1.1 g十六烷基三甲基溴化铵和0.48 g氢氧化钠的混合溶液中,搅拌均匀。再将1,2-双(三乙氧基硅基)乙烷1.6 mL分散于3 mL乙醇中,加入上述溶液中,室温搅拌反应30 min。将溶液转移到高压反应釜中,80 ℃下反应16 h。过滤收集白色沉淀,用大量乙醇洗涤,80 ℃下干燥,得到白色粉末状产物。再将1 g产物加入到200 mL含有1%(体积分数)浓盐酸的乙醇溶液中,50 ℃下搅拌6 h,去除模板剂,过滤,依次用大量水和乙醇洗涤,50 ℃下真空干燥6 h,即可得到β-环糊精/硅基杂化手性固定相。
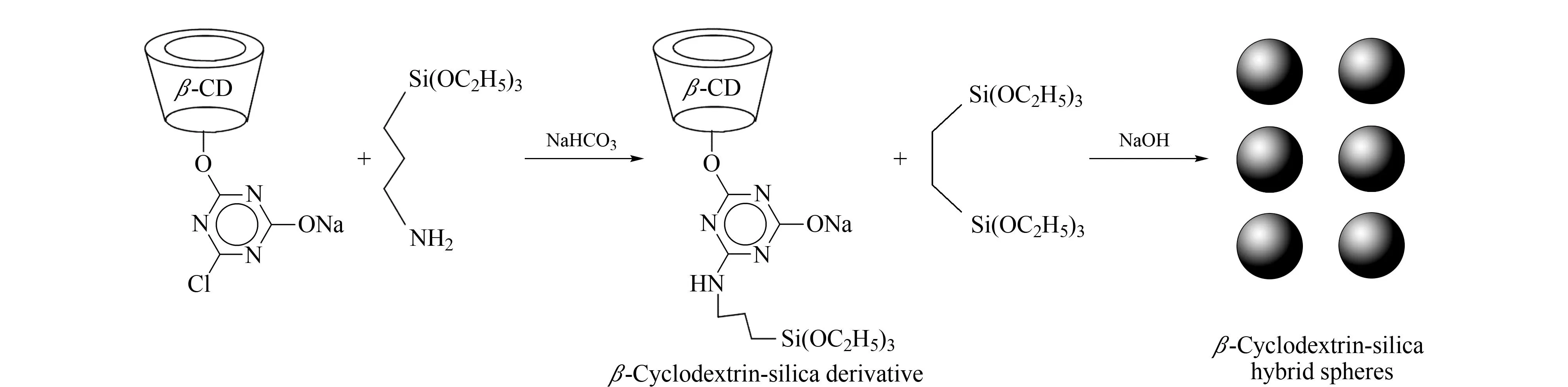
图1 β-环糊精/硅基杂化手性固定相的制备过程Fig. 1 Preparation of β-cyclodextrin (β-CD)/silica-based hybrid chiral stationary phase
1.4色谱柱的制备
以异丙醇为匀浆液,以甲醇为顶替液,在45 MPa的压力下,将固定相填充到不锈钢柱管中,即可制得β-环糊精/硅基杂化手性色谱柱。
1.5色谱计算
色谱柱死时间(t0)由丙酮测定[16];保留因子(k)按k1=(t1-t0)/t0,k2=(t2-t0)/t0计算,t1和t2分别为两个洗脱峰的出峰时间;分离因子α=k2/k1;分离度Rs=1.18(t1-t0)/(wt1+wt2),wt1和wt2分别为两个洗脱峰的半峰宽。

表 1 β-环糊精/硅基杂化手性固定相的手性分离
k1: retention factor 1;k2: retention factor 2;α: separation factor;Rs: chromatographic resolution. Mobile phase: Ⅰ, acetonitrile-1 % triethylammonium acetate (TEAA) (20∶80, v/v); Ⅱ, methanol-1%TEAA (35∶65, v/v).
1.6色谱条件
流动相以一定体积的乙腈/甲醇/三乙胺与醋酸缓冲盐(TEAA)配制(见表1),流速为0.6 mL/min;流动相均经超声波脱气后使用,每次更换流动相时要充分平衡色谱柱,所有的色谱分离均在室温下进行。样品使用流动相进行溶解,每次进样10 μL。检测器为紫外检测器,检测波长为250~280 nm。
2结果与讨论
2.1β-环糊精/硅基杂化手性固定相的制备
使用有机-无机杂化制备技术,通过两步化学反应制备了β-环糊精/硅基杂化材料。在制备过程中,乙烷功能基团分布于杂化材料的孔壁骨架中,而β-环糊精功能基团被模板剂包埋在孔道中,在脱除模板剂后,β-环糊精功能基团则留在孔道中,制备了具有介孔孔道的β-环糊精/硅基杂化手性固定相。本文制备的β-环糊精/硅基杂化手性固定相,不涉及环糊精衍生物的复杂合成反应和硅胶键合反应,β-环糊精功能基团在孔道中分布均匀,且负载量高于普通键合的手性固定相,不受硅胶表面硅羟基数量的限制,通过简单调节β-环糊精功能基团的比例可制备出不同负载量的β-环糊精/硅基杂化手性固定相。本方法制备过程简单、高效、成本低。
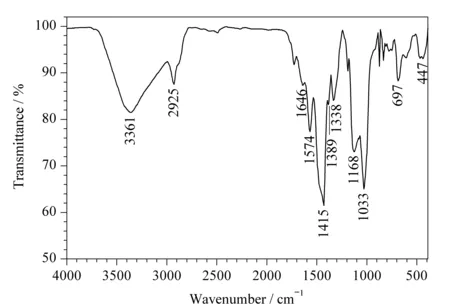
图2 β-环糊精硅基衍生物的红外光谱图Fig. 2 Infrared spectrum of β-cyclodextrin-silica derivative
2.2β-环糊精硅基衍生物的表征
由红外光谱图(图2)可以看出,在580 cm-1附近的C-Cl键的特征峰明显减小,1 646~1 415 cm-1处为三嗪环的振动特征峰,697 cm-1和1 033 cm-1处为Si-O键的红外振动特征峰,2 925 cm-1处为C-H键的伸缩振动特征峰,1 168 cm-1处为C-O键的伸缩振动特征峰,3 361 cm-1处为环糊精羟基振动特征峰。红外光谱表明β-环糊精硅基衍生物制备成功。
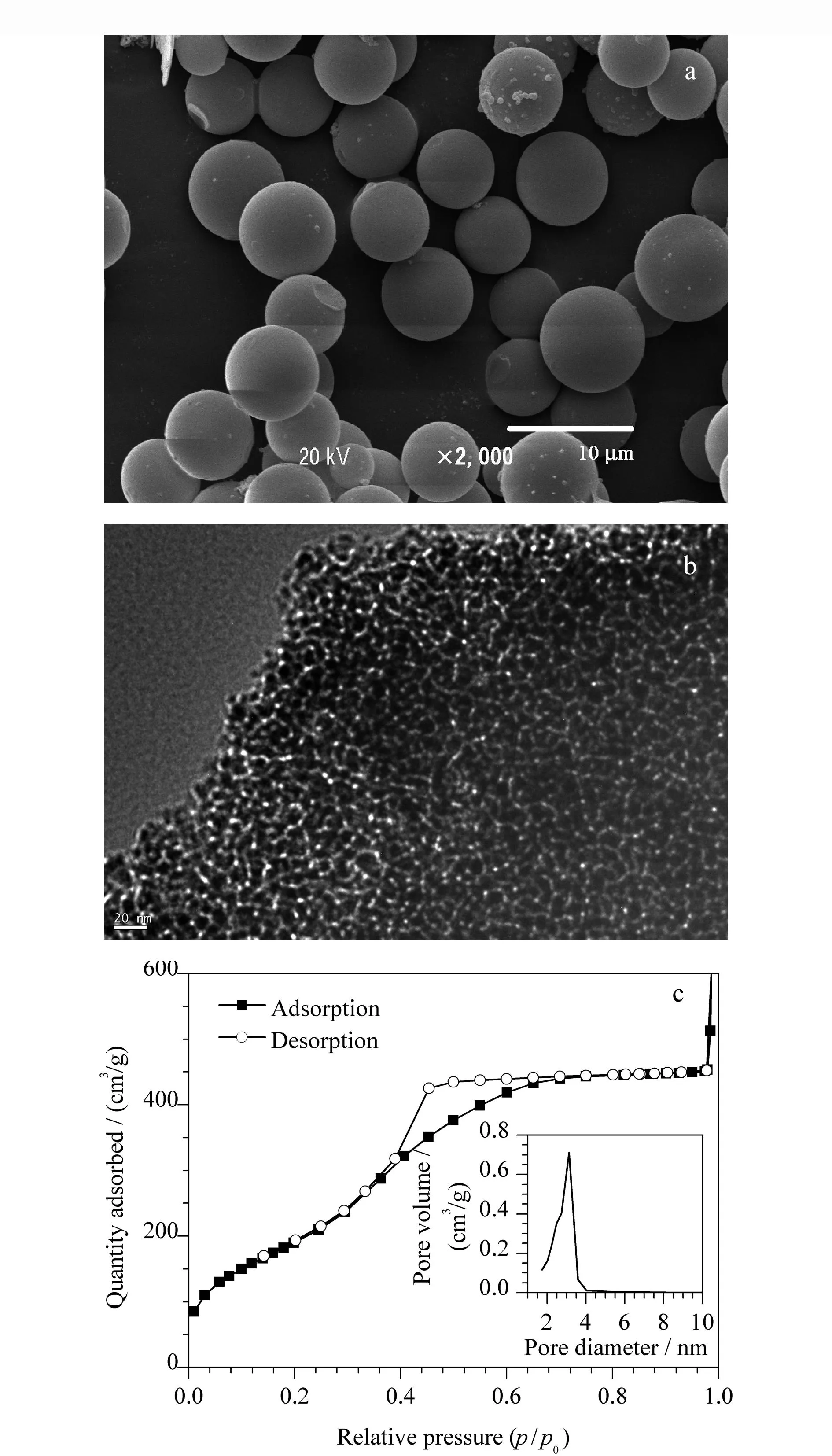
图3 β-环糊精/硅基杂化手性固定相的(a)扫描电镜图、 (b)高分辨率透射电镜图及(c)氮气吸附等温线和孔径分布图Fig. 3 (a) Scanning electron microscope micrograph, (b) high resolution transmission electron microscope micrograph and (c) nitrogen sorption-desorption isotherms and the pore size distribution of β-cyclodextrin/silica-based hybrid chiral stationary phase
2.3β-环糊精/硅基杂化手性固定相的表征
扫描电镜图(图3a)显示本文制备的β-环糊精/硅基杂化硅球的球形形貌规则,单分散性良好,硅球表面光滑,粒径为5~8 μm。
常规的色谱分离材料都具有良好的介孔孔道结构,本文制备的β-环糊精/硅基杂化硅球孔道结构的高分辨透射电镜图如图3b所示,硅球的边缘具有蠕虫状的孔道结构,孔道直径为2~4 nm。这些孔道可提高传质及增大吸附容量,有利于增强色谱材料的手性识别与分离。
图3c给出了β-环糊精/硅基杂化硅球的氮气吸附与解吸等温线和孔径分布图。从比表面孔度分析来看,制备的杂化硅球填料显示了典型的Ⅳ类吸附等温线和H1型滞后回线。Ⅳ类吸附等温线表明颗粒均匀,孔结构规则有序,孔径范围窄。H1型滞后回线表示孔结构为筒状中空结构,有利于样品分子的自由扩散。氮气吸附和脱附量p/p0在0.4至0.7间逐渐增大。从孔径分布图可看出大多数孔径在3.2 nm左右,与透射电镜得到的孔径数据基本吻合。氮气吸附脱附BET(Brunauer Emmett Teller)法测得的比表面积为816 m2/g,具有高的比表面积。
由图4a可知制备的β-环糊精/硅基杂化手性固定相的红外光谱中出现了2 922 cm-1及2 887 cm-1处的吸收峰,是骨架中1,2-亚乙基和环糊精中亚甲基的红外谱带。在693、919和1 040 cm-1处的吸收峰对应于Si-O键的红外振动特征峰,说明杂化材料中有硅烷基的存在。在3 465 cm-1处为羟基的伸缩振动特征峰,在1 643~1 412 cm-1处为三嗪环的振动特征吸收峰,在1 165 cm-1和1 105 cm-1处为C-O键的伸缩振动特征峰,这表明了β-环糊精功能基团的存在。图4b为β-环糊精/硅基杂化手性固定相的光电子能谱图,可以看到硅球材料具有C、N、O和Si 4种主要元素,进一步证明了本文制备的有机-无机杂化硅球材料成功地负载了β-环糊精功能基团。
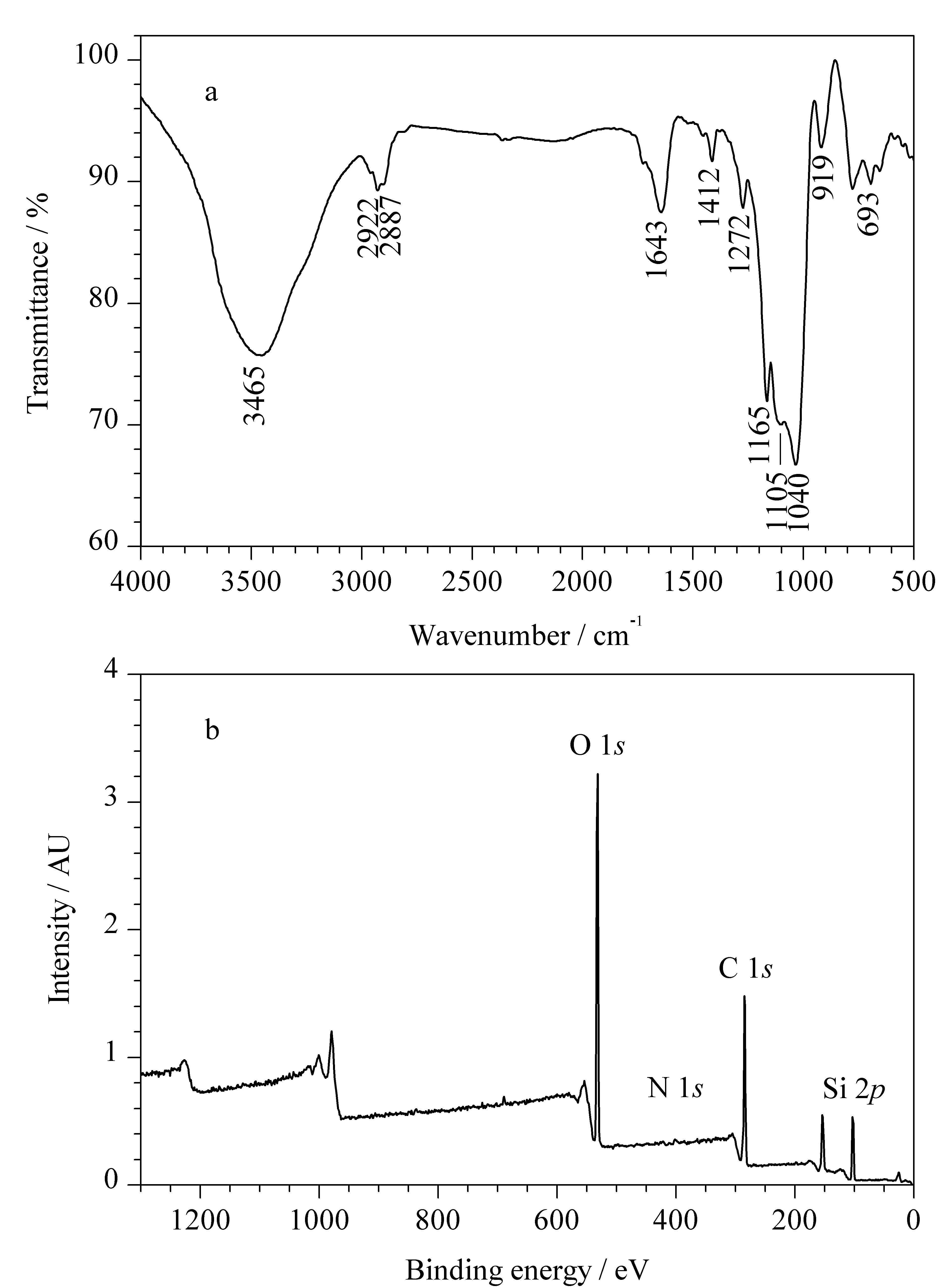
图4 β-环糊精/硅基杂化手性固定相的(a)红外光谱图和(b)光电子能谱图Fig. 4 (a) Infrared spectrogram and (b) X-ray photoelectron spectroscopy of β-cyclodextrin/silica-based hybrid chiral stationary phase
利用元素分析仪测得制备的β-环糊精/硅基杂化硅球的碳含量是17.33%,氮含量是0.75%,氢含量为3.99%。根据已经报道的键合量计算公式[17],由氮元素的含量可以计算出β-环糊精负载量为0.23 μmol/m2,并且本文制备的杂化硅球中β-环糊精的负载量明显高于传统的键合方法制备的环糊精手性固定相[18,19]。
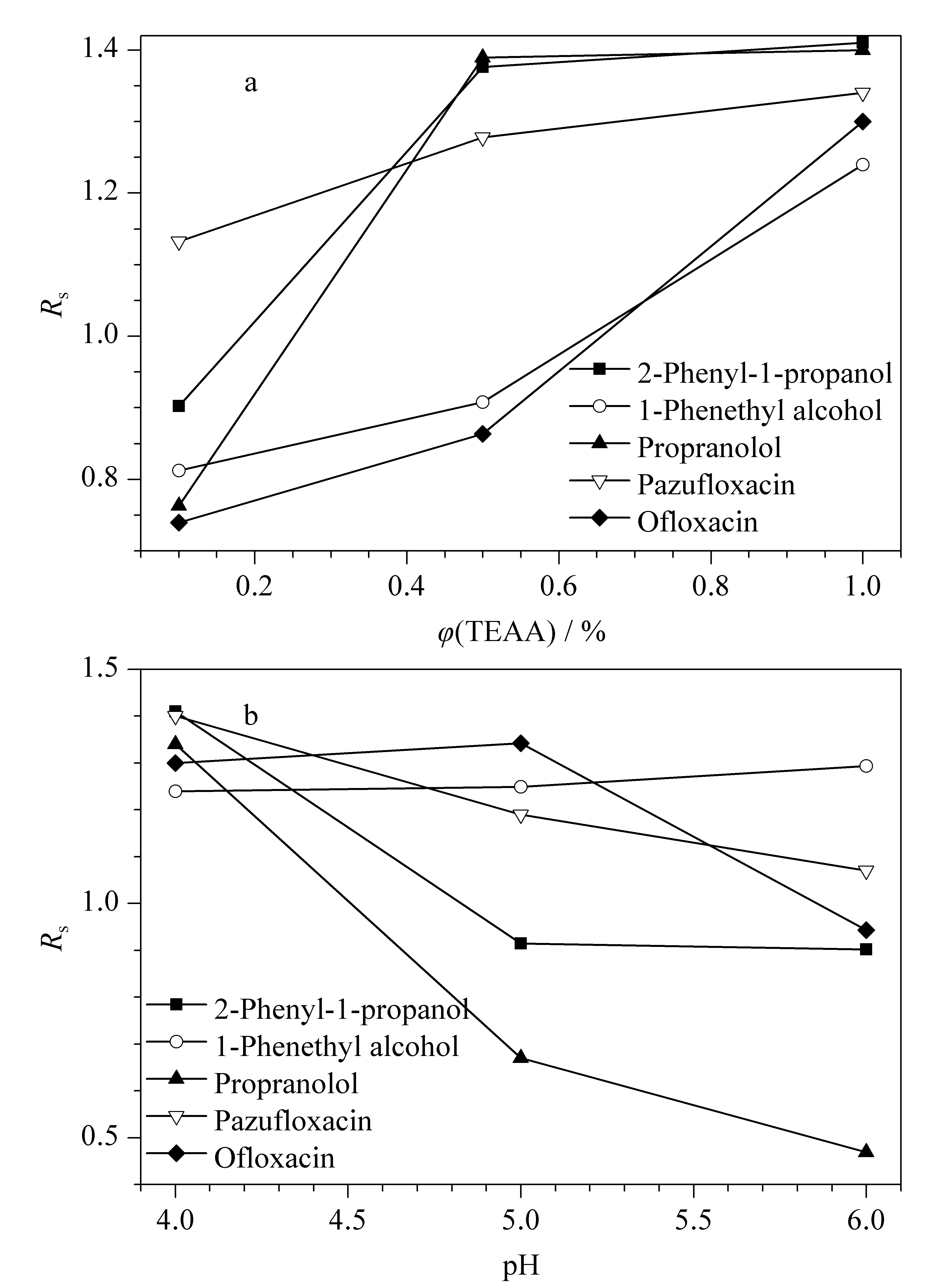
图5 (a)流动相中TEAA 体积分数和(b)pH对手性拆分的影响Fig. 5 Effects of (a) TEAA volume percentage and (b) pH value in mobile phase on the enantioseparations
2.4β-环糊精/硅基杂化手性固定相的手性拆分性能
2.4.1流动相组成对色谱分离的影响
在手性色谱分离中,缓冲液对取得理想的分离效果至关重要。流动相中加入缓冲液,一方面可以抑制溶质分子的离子化,改善其在色谱柱上的保留和选择性;另一方面也可以掩蔽或修饰色谱柱填料的极性基团,使色谱峰变得更规则,提高分离能力。缓冲液pH值的改变影响溶质分子功能基团与环糊精的相互作用或形成氢键的能力。本文考察了流动相中TEAA的体积分数及pH值对手性化合物拆分的影响。如图5所示,随着TEAA体积分数从0.1%升高到1%,保留时间减小,分离度增加,可能是因为浓度高的TEAA与环糊精形成氢键,从而使包合物的稳定性降低。随着pH值的增大,多数化合物的分离度降低。本实验选择pH值为4, TEAA体积分数为1%。
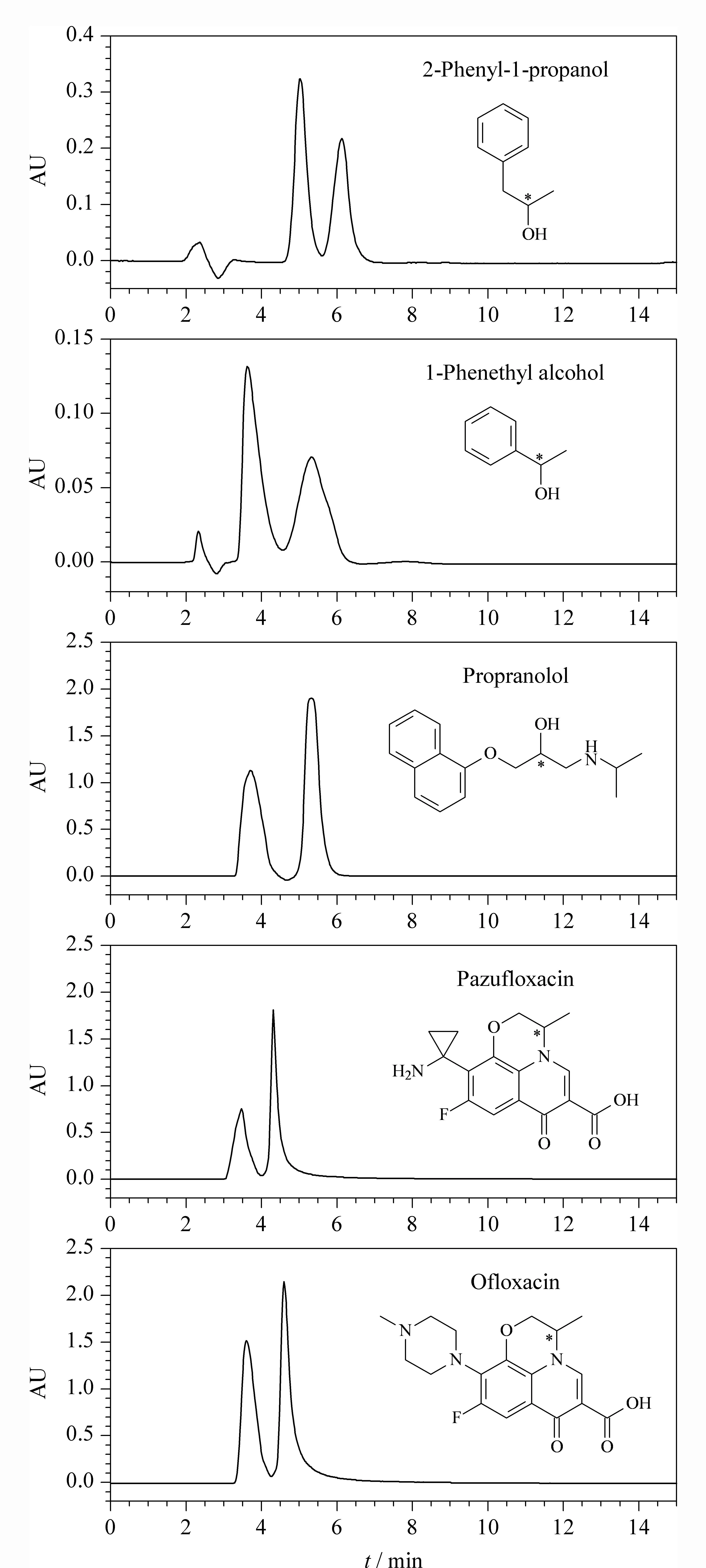
图6 手性化合物的拆分色谱图Fig. 6 Chromatograms of the enantioseparation of the chiral compounds
2.4.2手性拆分性能
由于环糊精本身的结构特点,其手性分子识别能力显著,被广泛地用于对映体的拆分。在环糊精分子中,糖单元都采用椅式构象,并且形成中空的圆锥体结构,水溶性和非水溶性化合物都能进入空穴形成包合物。客体分子能够全部或部分进入环糊精的空腔,主要通过客体与手性空腔的匹配作用进行手性识别和拆分分离,而苯环类化合物能够和环糊精的空腔进行很好的匹配,因此环糊精类手性固定相多用来分离带苯环的手性化合物。本文制备的β-环糊精/硅基杂化硅球材料用于色谱柱填料,在反相色谱条件下,采用乙腈/甲醇/TEAA作流动相,对2-苯基-1-丙醇、1-苯乙醇、普萘洛尔、甲磺酸帕苏沙星和甲磺酸氧氟沙星手性化合物进行拆分。对映体拆分色谱图如图6所示,5种手性化合物实现了基线分离。其色谱分离数据如表1所示,所拆分手性化合物的分离度都大于1,特别是2-苯基-1-丙醇的分离度为1.41,表明本文制备的孔道中负载β-环糊精杂化手性固定相具有很好的手性识别与拆分能力。主要归因于两方面:一方面,β-环糊精/硅基杂化硅球材料骨架的孔壁含有大量的亚乙基,在色谱分离过程中提供良好的疏水作用,在环糊精疏水性穴腔的包结作用过程中以协同作用的方式增强手性分离;另一方面,β-环糊精/硅基杂化硅球材料在孔道中的环糊精易裸露在流动相中,能与流动相中的溶质分子进行有效的手性识别作用。
与3,5-二甲基苯基衍生化的β-环糊精/硅基杂化手性固定相的拆分[15]比较,本文制备的手性固定相对2-苯基-1-丙醇、1-苯乙醇和普萘洛尔也实现了对映体拆分,但分离度有所降低,表明环糊精分子中的羟基被3,5-二甲基苯基衍生后有利于增强固定相的手性拆分能力。本文制备的杂化手性固定相对帕苏沙星和氧氟沙星具有较好的手性识别能力,表明在反相色谱条件下,未衍生的环糊精在外亲水和内疏水的包结作用下,容易识别这两种手性化合物。
3结论
本文合成了β-环糊精硅基衍生物,以十六烷基三甲基溴化铵为模板,在碱性条件下,β-环糊精硅基衍生物和1,2-双(三乙氧基硅基)乙烷发生聚合反应,制备了β-环糊精/硅基杂化硅球,利用红外光谱、扫描电镜、透射电镜、氮气吸附-脱附和元素分析等手段对其进行了表征,证明β-环糊精/硅基杂化硅球手性固定相制备成功。在没有对环糊精羟基进行衍生的情况下研究了其反相色谱手性分离性能,结果表明在手性物质分离过程中,其与流动相pH值及TEAA含量有较大关系;手性固定相对5种手性化合物实现了基线分离,表现出了较好的手性识别和拆分能力,在手性药物拆分方面具有较大的应用潜力。
参考文献:
[1]Lorenz H, Seidel-Morgenstern A. Angew Chem Int Ed, 2014, 53(5): 1218
[2]Tu H S, Fan J, Zhang W G, et al. Chinese Journal of Chromatography, 2014, 32(5): 452
涂鸿盛, 范军, 章伟光, 等. 色谱, 2014, 32(5): 452
[3]Xiao Y, Ng S C, Tan T T Y, et al. J Chromatogr A, 2012, 1269: 52
[4]Ward T J, Ward K D. Anal Chem, 2012, 84(2): 626
[5]Wang Y, Chen H, Xiao Y, et al. Nat Protoc, 2011, 6(7): 935
[6]Lai X, Tang W, Ng S C. J Chromatogr A, 2011, 1218: 5597
[7]Chen X, Zou H, Zhang Q, et al. J Chromatogr Sci, 2002, 40(6): 315
[8]Wang R Q, Ong T T, Ng S C. Tetrahedron Lett, 2012, 53(18): 2312
[9]Nicole L, Laberty-Robert C, Rozes L, et al. Nanoscale, 2014, 6(12): 6267
[10]Sanchez C, Shea K J, Kitagawa S. Chem Soc Rev, 2011, 40(2): 471
[11]Yang F, Mao J, Zhang Y K, et al. Chinese Journal of Chromatography, 2013, 31(6): 531
杨帆, 毛劼, 张玉奎, 等. 色谱, 2013, 31(6): 531
[12]Huq R, Mercier L, Kooyman P J. Chem Mater, 2001, 13(12): 4512
[13]Degoutin S, Bacquet M. J Porous Mater, 2013, 20(4): 663
[14]Zhang Z, Wu M, Wu R A, et al. Anal Chem, 2011, 83(9): 3616
[15]Wang L T, Dong S Q, Han F, et al. J Chromatogr A, 2015, 1383: 70
[16]Xiao X H, Liu X, Jiang S X. Chinese Journal of Chromatography, 2002, 22(1): 61
肖小华, 刘霞, 蒋生祥. 色谱, 2002, 22(1): 61
[17]Berthod A, Chang C D, Armstrong D W. Talanta, 1993, 40(9): 1367
[18]Lin C, Liu W, Fan J, et al. J Chromatogr A, 2013, 1283: 68
[19]Zhang Y, Guo Z, Ye J, et al. J Chromatogr A, 2008, 1191: 188
Preparation and enantioseparation performance of β-cyclodextrin-silica hybrid chiral stationary phases
WANG Litao1, DONG Shuqing1, ZHANG Zhixin1, WANG Yangjun2,ZHANG Xiaoli1, ZHANG Xia1, ZHANG Pengyun2,3, ZHAO Liang1*
(1. Key Laboratory of Chemistry of Northwestern Plant Resources and Key Laboratory for Natural Medicine of Gansu Province, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China;2. Northwest Normal University, Lanzhou 730000, China;3. Gansu Research Institute of Chemical Industry, Lanzhou 730000, China)
Abstract:A simple preparation method for β-cyclodextrin-silica hybrid chiral stationary phases was developed. Firstly, the β-cyclodextrin-silica derivative was synthesized by the reaction of 3-aminopropyltriethoxysilane and monochlorotriazinyl β-cyclodextrin under weak base condition. Spherical β-cyclodextrin-silica hybrid materials with β-cyclodextrin in the surface of pores by covalent bonding were prepared using 1,2-bis(triethoxysilyl) ethane and the β-cyclodextrin-silica derivative under the alkaline condition by one-step polymerization reaction. The β-Cyclodextrin-silica hybrid chiral stationary phases could be directly used as high performance liquid chromatographic packings after the template removal. The hybrid materials prepared in this paper possessed regular spherical morphology, good monodispersion, high specific surface area, good mechanical property, high chemical stability and simple preparation process. It combined the chiral recognition performance of β-cyclodextrin and the outstanding performance of organic-inorganic hybrid material. The effect of the composition, ratio and pH of mobile phase on chiral separation was investigated, and the best chiral separation conditions had been optimized. The baseline chiral separations for five chiral compounds were obtained under the optimal conditions. The results of enantioseparation showed that the hybrid chiral stationary phases had favorable chiral recognition ability. Compared with the traditional preparation process of chiral stationary phases, a new thought for new type of chiral stationary phase is provided by the present method in this paper.
Key words:high performance liquid chromatography (HPLC); chiral stationary phases; β-cyclodextrin; organic-inorganic hybrid; enantioseparation
DOI:10.3724/SP.J.1123.2015.06036
*收稿日期:2015-06-20
基金项目:国家自然科学基金项目(21405162,21405161);乌鲁木齐市科技局重点项目(Y141320007);阿拉善盟科技局沙产业科研项目.
中图分类号:O658
文献标识码:A
文章编号:1000-8713(2016)01-0089-07
色谱手性分离专刊·研究论文
*通讯联系人. Tel:(0931)4968261,E-mail:zhaol@licp.cas.cn.
Foundation item: National Natural Science Foundation of China (21405162, 21405161); Key Projects of Urumqi Science and Technology Bureau (Y141320007); Deserticulture Projects of Alxa League Science and Technology Bureau.
猜你喜欢
今日农业(2022年14期)2022-09-15
当代陕西(2019年23期)2020-01-06
中成药(2018年8期)2018-08-29
中成药(2018年4期)2018-04-26
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
科学与财富(2016年28期)2016-10-14
山东青年(2016年2期)2016-02-28
环境与生活(2016年6期)2016-02-27
化工进展(2015年2期)2015-08-19
