
Pseudomonas plecoglossicida NyZ12基因无痕敲除方法的建立
2016-06-13 10:44毛灵琪李存治闫达中
生物技术通报 2016年4期
毛灵琪 李存治 闫达中
(武汉轻工大学生物与制药工程学院,武汉 430023)
Pseudomonas plecoglossicida NyZ12基因无痕敲除方法的建立
毛灵琪 李存治 闫达中
(武汉轻工大学生物与制药工程学院,武汉 430023)
旨在建立一种适合环己胺降解菌NyZ12基因无痕敲除的可靠方法。通过overlapping PCR技术将目的基因上下游同源臂融合并克隆到自杀载体pEX18km上,将重组质粒转化到大肠杆菌S17 pir中,再通过接合转移到假单胞菌NyZ12菌株内,经pEX18km质粒上sacB基因的反向筛选得到突变株并通过PCR方法和测序鉴定。结果显示,成功构建了假单胞菌NyZ12菌株orf4637的基因突变株(NyZ12Δ4637)。通过自杀载体同源重组可以成功获得敲除的无痕突变株,且突变株基因组上没有任何抗性筛选标记残留,为环己胺降解菌NyZ12基因功能研究提供了可靠的基因敲除技术。
基因敲除;自杀载体;同源重组;结合转移
环己胺(cyclohexylamine,化学式C6H13N)又称六氢苯胺,是一种重要的精细化工中间体,用于生产金属缓蚀剂、杀菌杀虫剂、橡胶硫化促进剂和防老剂等。在生产和使用过程中,环己胺被释放到环境中,可通过呼吸和皮肤进入人体,实验已证实该化合物有致癌性[1]。由于环己胺在工业生产上的广泛应用和对人体的明显危害性,研究对环己胺有降解作用的微生物并了解其分子机制,是消除环己胺对环境污染和残留的有效途径之一。
目前报道的以环己胺为碳氮源生长的纯培养物仅有两株,一株是Iwaki 等[2]分离筛选到的革兰氏阳性菌Brevibacterium oxydans IH-35A,另一株是由中国科学院武汉病毒研究所周宁一[3]研究组分离的革兰氏阴性菌Pseudomonas plecoglossicida NyZ12。
NyZ12是一株能够利用环己胺作为唯一碳源、氮源生长的革兰氏阴性菌株,根据其16S rRNA基因序列分析为假单胞菌属,鉴定为变形假单胞菌Pseudomonas plecoglossicida NyZ12。这与Iwaki等分离筛选到能降解环己胺的革兰氏阳性短杆菌Brevibacterium oxydans IH-35A不同,也是目前为止国内第一次报道能降解环己胺的细菌。
本实验室对该菌株进行全基因组测序,获得全基因组信息[4]。通过对比分析,预测orf4637基因可能与环己胺降解过程有关。研究基因功能的重要方法之一是构建基因的突变体来检测其表型的变化,以推测某个基因在生长过程中起到何种作用。其中最直接的方法是将目的基因敲除,这样可以保证被研究的基因完全失活[5]。因此,为了进一步研究基因orf4637的功能,本研究采用无痕敲除技术构建Δ4637基因敲除体系。通过overlapping PCR技术[6]将目的基因上下游同源臂融合并克隆到自杀载体上,再将构建的敲除载体转移至受体菌,利用同源重组原理取代基因组上序列相同或非常相近的基因并使后者完全丧失功能[7]。旨在为后期对敲除突变体NyZ12Δ4637进行表型研究,确定其在生长中是否起关键作用奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 假单胞菌NyZ12(AmpR);E.coli S17 pir菌株(StrR);pGEM-T Easy载体(AmpR);自杀质粒pEX18km(KanR),所用抗生素浓度为氨苄青霉素Amp(100 g/mL),链霉素Str(10 g/L),卡那霉素Kan(50 g/L)。
1.1.2 主要试剂 5×TransStart FastPfu buffer、TransStart FastPfu DNA Polymerase、2.5 mmol/L dNTPs、限制性内切酶EcoR I和Sal I,T4 DNA Ligase均购自TaKaRa公司;质粒快速提取试剂盒、PCR产物凝胶回收试剂盒(离心柱型)、基因组提取试剂盒均购自上海捷瑞生物工程有限公司。
CCMB80溶 液:KAc(1 mol/L pH8.0)1 mL,0.891 g NaCl,Glycerol 10 mL,MnCl2·4H2O(1 mol/L) 1 mL,MgCl2·6H2O(1 mol/L)1 mL,定容至100 mL,过滤除菌,冰上储存备用。
LB培养基:NaCl 1 g,Tryptone 1 g,Yeast Extract 0.5 g,加水定容至100 mL,121℃下高温灭菌20 min。
环己胺无机盐培养基:KH2PO41.5 g,Na2HPO4· 12H2O 3.8 g,定容至1 L,121℃高温灭菌;再依次加入高温灭菌的10 mL 1 g/L CaCl2·2H2O;10 mL过滤除菌的5 g/L MgSO4·7H2O、0.2 g/L MnSO4·4H2O及0.5 g/L FeSO4·7H2O的混合体系,10 mL过滤除菌1 mol/L的环己胺盐酸盐溶液(pH7)。
RGMC:1% Tryptone;0.1% Yeast extract;0.8% NaCl;0.1% glucose;5 mmol/L MgCl2;5 mmol/L CaCl2。其他生化试剂均从国药集团化学试剂有限公司购买的分析纯试剂。
1.2 方法
1.2.1 同源臂扩增 设计两对引物分别扩增orf4637基因上游约700 bp及下游约900 bp大小同源臂,引物序列见表1。以环己胺降解菌NyZ12基因组DNA作为模板,引物对分别为P1/P2和P3/P4进行PCR反应,PCR体系如下:模板0.3 μL,FastPfu酶1 μL,5×FastPfu buffer 10 μL,上下游引物各1 μL,加无菌去离子水至50 μL。扩增程序为:98℃ 2 min,98℃ 20 s,60℃ 20 s,72℃ 20 s,30个循环;72℃ 5 min。凝胶电泳回收PCR产物。

表1 用于 4637突变株构建及鉴定的引物
1.2.2 融合PCR反应 纯化后的同源臂片段1∶1混合,将混合DNA片段作为模板进行融合PCR,引物对为P1/P4。PCR体系和扩增程序同上,延伸时间延长至1 min,将纯化PCR产物加“A”后克隆到pGEM-T Easy载体上,酶切鉴定连接正确大小片段的克隆并测序。
1.2.3 缺失突变载体的构建 分别以EcoR I和Sal I对pEX18km质粒和连有目的片段的pGEM-T Easy载体进行双酶切并经过凝胶电泳回收。将酶切后的质粒和目的片段按适当的比例混合,加入T4连接酶在16℃连接过夜,再将连接产物转化到E.coli S17 pir感受态细胞中,转化产物在37℃,180 r/min的LB培养基中活化1 h后,取100 μL涂布于LB平板上(StrR,KanR),37℃培养至出现单菌落。通过酶切鉴定筛选出含有融合片段的重组质粒。
1.2.4 结合转移[8]和单交换 将含重组质粒的E.coil S17 pir接种在LB培养基中,环己胺降解菌NyZ12接种在无机盐培养基中。分别在37℃和28℃,170 r/min培养至OD600=0.5-0.8。随后以供体菌(含有重组质粒E.coil S17 pir)与受体菌(NyZ12)1∶2和1∶5的比例混合菌液,6 000 r/min离心30 s收集菌体,用100 μL RGMC将菌体洗涤两遍,最后50 μL重新悬浮并滴加在滤膜上。待菌液被滤膜充分吸收后,将其案贴在RGMC平板上,28℃培养8-12 h。之后用100 μL无菌水洗涤滤膜,洗下的菌液涂布在含有氨苄青霉素和卡那霉素的双抗LB平板上,28℃培养至长出单克隆菌落。根据抗性筛选和PCR鉴定,找到发生单交换菌株。
1.2.5 双交换 挑单交换菌株接种于LB培养基中(AmpR),28℃,170 r/min多次传代培养。传至第4代将菌液稀释100倍后取100 μL涂布于10%蔗糖的LB平板上,28℃培养至出现单菌落。挑取单克隆接种于含有Amp的LB培养基中培养,再将过夜培养的菌液取4 μL分别接种于含有Amp或者含有Kan和Amp双抗LB培养基中,28℃培养。挑选能在Amp培养基中生长但不能在双抗培养基中生长的单克隆进行PCR鉴定,鉴定引物P1/P4和E1/E2。
将本实验中突变载体的构建和交换过程分别总结为图1、图2。
1.2.6 测序验证 煮沸法[9]提取敲除菌的基因组,并以敲除菌基因组DNA作为模板,引物对分别为P1/E2进行PCR反应,PCR体系如下:模板4 μL,FastPfu酶1 μL,5×FastPfu buffer 10 μL,引物各1 μL,加无菌去离子水至50 μL。扩增程序为:98℃ 2 min;98℃ 20 s,58℃ 20 s,72℃ 30 s,30个循环;72℃ 5 min。凝胶电泳回收PCR产物并送至生工测序部测序。
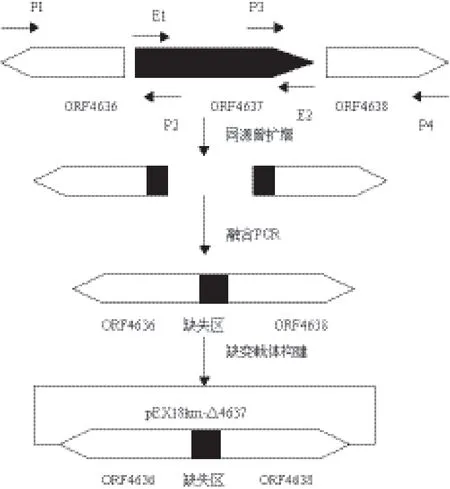
图1 Δ4637突变载体构建步骤流程图

图2 单交换和双交换过程
1.2.7 生长实验 将敲除菌与野生菌按0.5%的接种量接种于装有50 mL无机盐培养基的三角瓶(250 mL)中,从12 h开始取样,然后每隔1 h测定菌液OD值,绘制敲除菌与野生菌的生长曲线,比较生长状况。
2 结果
2.1 同源臂与融合PCR
扩增引物对P1/P2扩增orf4637上游同源臂(约663 bp),引物对P3/P4扩增orf4637下游同源臂(约863 bp),成功融合的片段大小为1 526 bp。1%琼脂糖电泳结果(图3、图4)显示,PCR产物大小与预期相符,通过凝胶电泳回收得到高纯度的目的片段。
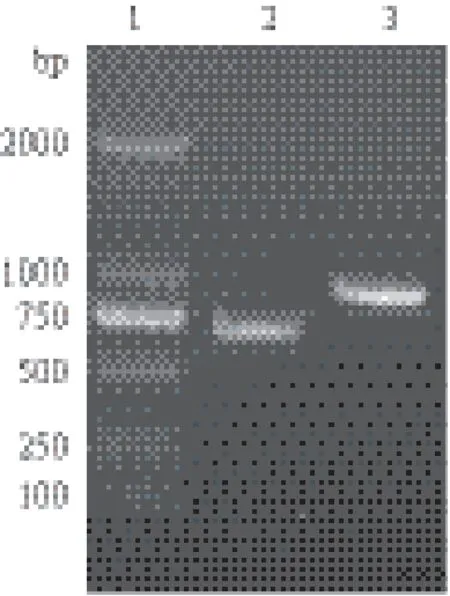
图3 PCR扩增同源臂

图4 PCR扩增融合片段产物
2.2 重组质粒的构建
将融合片段加A后连接到pGEM-T Easy载体上,抽取质粒并酶切鉴定,确定目的片段连接T载体上的阳性克隆测序。提取质粒pEX18km并用EcoR I和Sal I进行酶切,酶切后的DNA片段进行琼指糖电泳,结果(图5)显示,大小约为 6 100 bp,与标准序列大小相符。再用EcoR I和Sal I对连接目的片段的T载体片段进行双酶切,将两个片段分别回收后连接构建重组质粒pEx18km-△4637,转化至E.coli S17 pir菌株中,抽取质粒酶切鉴定,鉴定结果见图6。
2.3 单交换突变体的鉴定
E.coli S17 pir与环己胺降解菌通过微孔滤膜进行结合转移的效率比较高,重复性好。一般能在卡那霉素和氨苄青霉素抗性平板上生长的单克隆都是发生了单交换的阳性重组克隆。再挑取单菌落进一步鉴定,分别用扩目的基因orf4637的引物对E1/E2和基因敲除引物对P1/P4进行PCR鉴定。用引物E1/E2扩增有1 600 bp和120 bp左右两条DNA片段(图7,1、2泳道),以P1/P4扩增有3 000 bp和1 600 bp左右两条片段(图8,2、3泳道)的克隆就是发生单交换的突变株。

图5 pEx18km酶切后电泳图

图6 重组质粒pEx18km-△4637酶切鉴定图
2.4 双交换与突变株鉴定
单交换菌株在LB培养基中传4代,稀释后涂布于含有Amp和10%蔗糖的LB平板上。由于自杀质粒pEx18km上sacB基因突变率高,在平板上长出的单克隆也可能是单交换菌。但是双交换菌不具备Kan抗性,单交换菌具有Kan抗性,所以利用抗性筛选,找到可能的双交换突变体。再以该菌基因组为模板,分别用扩目的基因orf4637的引物对E1/E2和基因敲除引物对P1/P4进行PCR鉴定,鉴定结果如图9和图10所示。以引物E1/E2扩增只有120 bp大小片段(图9,1泳道),而外侧引物对P1/P4扩增只有1 600 bp左右片段的克隆即为发生双交换的阳性克隆,相对于单交换,无3 000 bp左右的扩增产物(图10,1泳道),其扩增产物比以野生株为模板的扩增产物小了约1 400 bp,即缺失了基因编码区。

图7 E1/E2引物对鉴定单交换

图8 P1/P4引物对鉴定单交换

图9 E1/E2引物对鉴定双交换

图10 P1/P4引物对鉴定双交换
2.5 敲除菌株测序结果
煮沸法提取敲除菌株基因组并利用引物P1/E2扩增缺失区得到约750 bp大小片段,如图11所示。
将目的片段凝胶电泳回收并送至生工测序部测序,结果如图12所示。

图11P1/E2引物扩敲除片段
由于这段序列靠近测序3'端,有几个碱基突变和缺失,可能是测序误差。这段序列已经跨过敲除区域,可以从两段序列的对比发现orf4637基因中间缺失,说明的确发生了双交换,orf4637成功敲除。
2.6 敲除菌生长实验
通过对敲除菌的生长实验的研究,绘制了敲除菌与野生菌的生长曲线(图13),比较发现敲除orf4637突变体相对野生型在环己胺的无机盐培养基中的生长变慢,但不是很明显。
3 讨论
目前国际上对环己胺的微生物降解研究并不多,微生物代谢环己胺的分子机理尚未揭示,因此以环己胺降解菌NyZ12为实验对象,构建一种可行的无痕敲除体系,成功敲除野生菌中的目的基因,对后续研究该菌的环己胺降解分子机制至关重要。利用同源重组技术敲除目的基因是目前研究基因功能最常用的定点突变手段,无痕敲除的主要优势是不需要在同源序列中插入抗性标记基因,这样不仅简化了敲除载体的构建,而且减少了插入抗生素基因产生极性效应对菌株生长产生的不良影响。同时构建敲除载体时,SacB 基因的引入使得无痕敲除效率大大提高。SacB基因来源于枯草芽孢杆菌,编码分泌性的蔗糖-6-果糖基转移酶,该酶催化蔗糖的水解和果聚糖的合成。在革兰氏阴性菌中,在有蔗糖存在的情况下,基因的表达对细菌来说是致死性的[10]。

图12 测序鉴定双交换突变体

图13 敲除菌的生长曲线
自杀载体pEx18km[11]不仅有反向筛选基因sacB还有Kan标记抗性,因此发生第一次交换后,将突变株涂布于含有氨苄青霉素和卡那霉素抗性的LB平板,因为野生菌本身具有Amp抗性而E.coli S17不含Amp抗性,因此能在平板上生长的单克隆,应该是整合了pEx18km的环己胺降解菌,也就是发生了单交换,再利用PCR方法确定,可以找到发生单交换的阳性克隆。此方法大大加快了构建缺失株的步伐,成为整个构建无痕突变株过程的关键步骤。再将多次传代的单交换菌株,涂布于高蔗糖浓度的LB平板。利用蔗糖的选择性压力和细菌染色体复制过程中发生的自由重组去除载体序列,产生无标记突变株[12,13]。在整合了pEx18km的环己胺降解菌中只有发生第二次交换的突变株才能在含有蔗糖的培养基中生长。抗生素抗性和sacB基因表达可作为正和负选择标记,但是自发的抗生素抗性、高异常重组率和基因突变可能会导致较高的假阳性,所以最后应对大量突变株进行鉴定[14]。
我们完成了环己胺降解菌NyZ12的基因组测序,预测基因组有5个胺氧化酶基因,最直接验证基因功能的方法是克隆表达基因,测定是否有酶活力。初步实验结果表明orf4637对底物环己胺有酶活力。通过基因敲除,观察突变体的生长情况,从另一个角度回答orf4637是否参与环己胺的代谢。对NyZ12Δ4637敲除菌株进行了生长实验,与野生菌对比,发现基本生长情况变化不明显,所以推断环己胺代谢途径中催化其第一步的环己胺氧化酶可能有多个,orf4637不是主效基因。这为后续逐一敲除其他胺氧化酶基因,研究突变体的表型奠定了基础。
4 结论
本研究选择可能与环己胺降解有关的orf4637作为目的基因,选用自杀质粒pEx18km作为载体,建立了成熟的环己胺降解菌NyZ12基因无痕敲除方法,成功构建了假单胞菌NyZ12Δ4637基因突变株。
[1]Bopp BA, Sonders RC, Kesterson JW. Toxicological aspects of cyclamate and cyclohexylamine[J]. Crit Rev Toxicol, 1986, 16:213-306.
[2]Iwaki H, Shimizu M, Tokuyama T, et al. Biodegradation of cyclohexylamine by Brevibacterium oxydans IH-35A[J]. Appl Environ Microb, 1999, 65:2232-2234.
[3]Shen Y, Yan DZ, Chi XQ, et al. Degradation of cyclohexylamine by a new isolate of Pseudomonas plecoglossicida[J]. World J Microbiol Biotechnol, 2008, 24:1623-1625.
[4]Li X, Li CZ, Mao LQ, et al. Complete genome sequence of the cyclohexylamine-degrading Pseudomonas plecoglossicida NyZ12[J]. Journal of Biotechnol, 2015, 199:29-30.
[5] 于慧敏, 马玉超. 工业微生物代谢途径调控的基因敲除策略[J].微生物代谢工程, 2010, 26(9):1199-1208.
[6]Lee J, Shin MK, Ryu DK, et al. Insertion and deletion mutagenesis by overlap extension PCR[J]. Methods in Molecular Biology, 2010, 634:137-146.
[7]黄勇. 细菌基因突变的策略及应用术[J]. 微生物学通报, 2007, 34(1):169-172.
[8]Simon RU, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering:transposon mutagenesis in Gram-negative bacteria[J]. Nat Biotech, 1983, 1:784-791.
[9]张志鸿, 甘蓓, 谭强来, 等. 基于不同靶基因的荧光定量PCR快速检测蜡样芽孢杆菌的研究[J]. 食品工业科技, 2013, 23(34):160-163.
[10]Hu F, Jiang X, Zhang JJ, et al. Construction of an engineered strain capable of degrading two isomeric nitrophenols via a sacB - and gfp -based markerless integration system[J]. Appl Microbiol Biotechnol, 2014, 98(10):4749-4756.
[11]Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, et al. A broadhost-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences:application for isolation of unmarked Pseudomonas aeruginosa mutants[J]. Gene, 1998, 212(1):77-86.
[12]Heng C, Chen ZJ, et al. Expression and secretion of an acid-stable α -amylase gene in Bacillus Subtilis by SacB promoter and signal peptide[J]. Biotechnol Lett, 2005, 27(21):1731-1737.
[13]Sun XY, Yang DD, Wang YY, et al. Development of a markerless gene deletion system for Streptococcus zooepidemicus:functional characterization of hyaluronan synthase gene[J]. Appl Microbiol Biotechnol, 2013, 97:8629-8636.
[14]Zhou QM, Fan DJ, Xie JB, et al. A method for generating precise gene deletions and insertions in Escherichia coli[J]. World J Microbiol Biotechnol, 2010, 26:1323-1329.
(责任编辑 李楠)
A Method for Constructing Unmarked Deletion Mutants of Pseudomonas plecoglossicida NyZ12
MAO Ling-qi LI Cun-zhi YAN Da-zhong
(School of Biology and Pharmaceutical Engineering,Wuhan Polytechnic University,Wuhan 430023)
This study aims to establish a practical method for an unmarked gene knockout of Pseudomonas plecoglossicida NyZ12 that degrades cyclohexylamine,which is significant for further studying the molecular mechanism of it degrading cyclohexylamine. The upstream and downstream flanking DNA fragments of the target gene were fused by overlapping PCR,and then cloned into the suicide vector pEx18km. The recombinant plasmid was introduced into Escherichia coli strain S17 pir and then transferred into NyZ12 by conjugation. The mutants by inverse screening sacB gene in pEx18km was identified by PCR and sequencing. The mutant NyZ12Δ4637 from NyZ12 strain orf4637 were successfully constructed. In conclusion,the unmarked gene-knockout mutant was acquired using homologous recombination of suicide vector,and there was no resistance residual of screening marker on the genome of the mutant. This study provides a reliable gene-knockout technology for investigating the genetic functions of NyZ12.
gene knockout;suicide vector;homologous recombination;conjugative transfer
10.13560/j.cnki.biotech.bull.1985.2016.04.027
2015-08-25
国家自然科学基金项目(31270112),微生物代谢国家重点实验室(上海交通大学)开放课题(MMLKF14-06)
毛灵琪,女,硕士研究生,研究方向:微生物学;E-mail:qzjsmlq@163.com
闫达中,男,博士,副教授,研究方向:微生物学;E-mail:yandz6808@163.com
猜你喜欢
今日农业(2021年11期)2021-08-13
空间科学学报(2021年1期)2021-05-22
食品科学(2018年10期)2018-05-23
环境保护与循环经济(2017年5期)2018-01-22
中国果菜(2016年9期)2016-03-01
中国蔬菜(2015年9期)2015-12-21
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
遗传(2014年3期)2014-02-28
