
Pt18团簇催化肉桂醛选择性加氢反应机理研究
2016-06-05 15:00李来才
四川师范大学学报(自然科学版) 2016年5期
魏 维,王 薇,李来才
Pt18团簇催化肉桂醛选择性加氢反应机理研究
魏 维,王 薇,李来才*
(四川师范大学化学与材料科学学院,四川成都610066)
采用密度泛函理论(DFT)中的B3LYP方法,在6-31+G(d,p)基组水平上,重点研究Pt18团簇对肉桂醛选择性加氢反应的催化作用.对反应通道上反应物、中间体、过渡态和产物进行结构优化.通过能量和振动频率分析以及内禀反应坐标(IRC)计算证实了过渡态和中间体的合理性.结果表明:Pt18团簇催化肉桂醛选择性加氢反应有6条不同的反应路径,其中,C——O键加氢产物的最低活化能为98.42 kJ/mol;C——C键加氢产物的最低活化能为122.88 kJ/mol.综合分析发现Pt18团簇催化肉桂醛选择性加氢反应中C——O键加氢所需的活化能最低.
密度泛函理论;Pt团簇;肉桂醛;选择性加氢反应
肉桂醛在精细化工、制药工业及食品工业应用广泛,其选择性加氢反应是一类十分重要的反应.肉桂醛分子中含有共轭的C——C、C——O键,其催化加氢反应可生成3-苯基丙烯醇、3-苯基丙醛及肉桂醇,肉桂醇继续加氢则生成3-苯基丙醇.在这些加氢产物中,肉桂醇的用途广泛,常用作制药中间体、定香剂和修饰剂.肉桂醛的选择性加氢反应越来越受到研究者的关注,提高其催化加氢反应的选择性,无论是在理论研究领域还是工业应用方面都具有重大意义.N.Mahata等[1]制备了以介孔碳为载体,分别用掺杂了Fe、Zn的Pt催化剂来研究肉桂醛的选择性加氢反应,结果表明:把Fe、Zn添加到Pt催化剂中,不仅提高了催化剂的活性,而且提高了向肉桂醇转化的选择性.同时,介孔碳的特殊结构也有利于提高催化剂的催化活性.高雪霞等[2]采用液相还原法制备了4种炭材料(分别是活性炭、碳纳米纤维、碳纳米管、碳气凝胶)负载的Pt催化剂,分别研究了这几种催化剂的活性及对肉桂醛加氢反应的选择性,结果表明:Pt粒子均匀分布在碳材料的表面,其中Pt/碳纳米纤维、Pt/碳纳米管都有助于C——O键活化加氢,并且Pt/碳纳米管的催化活性最高.A.K.Prashar等[3]报道了Pt/介孔SiO2催化肉桂醛加氢生成肉桂醇的研究,详细讨论了催化剂Pt的粒径大小对加氢选择性的影响.张付利等[4]用硼氢化钾还原肉桂醛合成了肉桂醇,研究了反应物的剂量、反应时间、温度等因素对肉桂醇产率的影响,结果表明:在室温下,当肉桂醛与硼氢化钾的物质的量之比为3∶1时,反应1.5 h后,肉桂醇的产率达到92%.在众多制备肉桂醇的方法中,通过肉桂醛的选择性催化加氢反应来制备肉桂醇得到了广泛应用和发展.由于金属催化剂拥有较高的选择性和催化活性,使其在催化加氢反应中发挥了巨大的作用.G.Neri等[5]在30℃、0.414 MPa的条件下,用Pt催化巴豆醛选择性加氢,使巴豆醛的转化率达到 40%,生成巴豆醇的选择性达到90%.S.Handjani等[6]制备了以 Al-SBA-15、SBA-15、Al2O3为载体的Pt催化剂,来催化肉桂醛选择性加氢,实验结果表明,Pt/Al2O3有利于C——O键加氢,而Pt/SBA-15、Pt/Al-SBA-15则有利于C——C键加氢.J.J.Shi等[7]用Pt催化肉桂醛选择性加氢,使其在十分温和的反应条件下生成了肉桂醇,且肉桂醛的转化率达97.8%,生成肉桂醇的选择性则高达85.3%.V.Gutiérrez等[8]在液相条件下,研究了Cu纳米催化剂催化α,β-不饱和醛的选择性加氢情况,发现反应的转化率及生成相应醇的选择性都较高.从大量的文献资料中发现,Pt是众多金属催化剂中催化活性和选择性都较高的一种金属,因此在催化选择性加氢反应中有广泛的应用.近年来,理论计算方法被广泛运用于催化剂优化反应微观机理的研究.它对反应机理的描述和寻找新方法通道有着独特的优势,能很好地解释催化剂的作用机制和诸多实验现象,并为实验结果提供可靠的理论数据.毛华平等[9]采用密度泛函理论优化并得到了Yn(n=2~8)小团簇的基态平衡结构,计算出了团簇的原子化能.结果表明,钇原子之间形成团簇最稳定的结构是倾向于平均配位数最大的结构.张萍等[10]在相对论有效原子实势近似的情况下,用密度泛函方法计算了掺杂二元合金PdnAu(n=1~5)团簇可能的几何构型和对应的电子态.结果表明,Pd-Au相互作用较强,改变了纯钯团簇的稳定构型,合金团簇的稳定性随体积的增大而降低.肖培等[11]采用生长法和替代法相结合的方式构建AlnTi2团簇可能的候选构型,并在密度泛函理论基础上对其优化得到一系列各尺寸团簇的平衡几何结构.结构表明,通过在Aln+1Ti团簇结构上替代一个Ti原子的方式所构成的模型占据了AlnTi2团簇稳定结构的主导地位.B.Kalita等[12]采用密度泛函理论分别研究了CO吸附在电中性团簇和带电荷的Pdn(n=1~7)团簇上的情况,得到了CO在Pdn(n=1~7)团簇上的稳定吸附构型.J.P.Chou等[13]采用密度泛函理论中的3种不同交换相关泛函研究了44种13个原子的金属团簇,发现广义梯度近似(GGA)与局域密度泛函近似(LDA)2种方法的计算结果一致.P.E.Balbuena等[14]研究了H2O2在Pt团簇和Pt-M(M=Cr,Co,Ni)团簇合金上的吸附和解离情况,得到了相关物质的吸附能和解离能等数据及稳定吸附构型.S.Laref等[15]采用密度泛函理论分别讨论了巴豆醛和丙烯醛在Pt(111)表面选择性加氢的情况,得到了优化后的构型、键长及能量等信息,结果表明生成饱和醛和饱和醇的选择性受动力学控制,生成肉桂醇的选择性则受热力学吸附控制.目前,关于Ptn团簇催化肉桂醛选择性加氢反应的机理研究并不多,为了深入了解Ptn团簇的催化作用本质,本文采用密度泛函理论重点探讨Pt18团簇催化肉桂醛选择性加氢反应的微观反应机理.
1 计算方法
本文采用密度泛函理论中的B3LYP方法,在6-31+G*(Pt采用赝势基组Lanl2dz[16])基组水平上对Pt18团簇催化肉桂醛选择性加氢反应体系中的所有物质进行几何结构优化,并在同一基组水平上进行频率计算和NBO[17]分析,对优化后各化合物的构型在B3LYP/6-311++G(d,p)计算水平上进行单点能计算和零点能矫正.通过内禀反应坐标(IRC)计算和振动频率分析进一步证实了过渡态的合理性.以上计算均采用Gaussian 09程序[18]完成.体系中各物质的BCP和RCP电荷密度由AIM 2000程序包[19]计算得到.
2 结果与讨论
2.1 Pt18团簇催化肉桂醛选择性加氢反应机理研究 研究表明Pt18团簇催化肉桂醛选择性加氢反应有6条不同的反应路径,具体反应历程见图1.
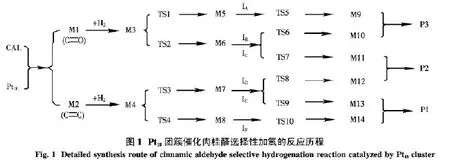
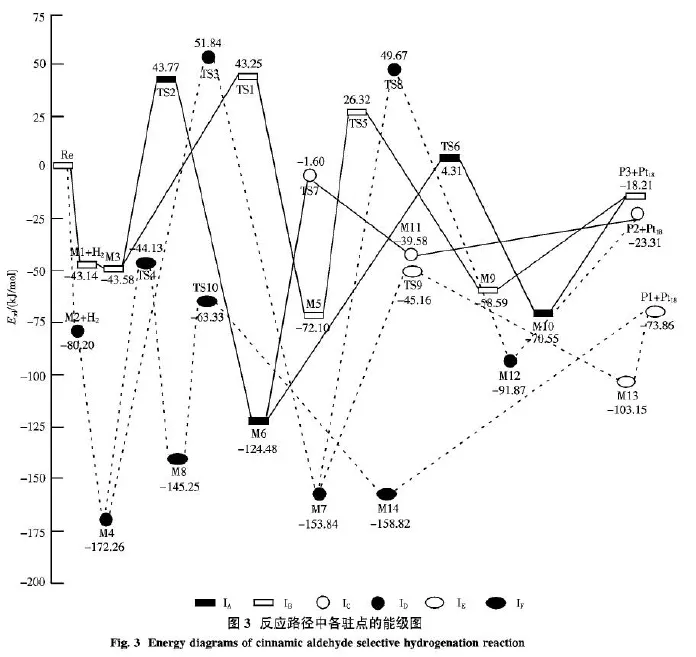
Pt18团簇分别与肉桂醛(Re)中的C——O、C——C键络合,形成中间体M1和M2,中间体分别与H2结合形成新的中间体M3和M4,经过一系列中间体和过渡态得到产物3-苯基丙醛(P1)、3-苯基丙烯醇(P2)和肉桂醇(P3).反应过程中所有物质的优化构型和结构参数见图2,相关能量列入表1,反应能级示意图见图3.

表1 反应各驻点的能量、相对能量和振动频率Table 1 Energies,relative energies and frequencies of compounds in reactions
选取最稳定的Pt18团簇构型作为催化剂模型(见图2),其中Pt(1)—Pt(2)键长为0.257 7 nm.第一步是Pt18团簇分别与肉桂醛中的C(4)——O、C(2)——C(3)键络合,形成中间体M1和M2.反应物Re中 C(4)——O、C(2)——C(3)键长分别为0.122 2和 0.135 3 nm,BCP电荷密度分别为0.397 7和0.336 5 a.u..中间体M1中C(4)——O键长为0.124 7 nm,BCP电荷密度为0.371 6 a.u..中间体M2中C(2)——C(3)键长为0.140 6 nm,BCP电荷密度为0.304 6 a.u..以上数据表明,肉桂醛与Pt18团簇活化络合后,C(4)——O、C(2)——C(3)键被削弱.由表1能量数据可知,Re+Pt18→M1、Re+ Pt18→M2过程中能量分别降低43.14和80.20 kJ/ mol,说明中间体M1、M2易形成并能稳定存在.由NBO计算分析可知,中间体M1中LP(2)Pt(1)→BD*(2)C(4)—O、BD(1)C(4)—O→LP*(6) Pt(1)的二阶稳定化能E(2)分别为7.79和7.07 kJ/mol,中间体 M2中 LP(5)Pt(1)→BD*(2) C(2)—C(3)、BD(2)C(2)—C(3)→LP*(6) Pt(1)LP(5)Pt(1)的二阶稳定化能E(2)分别为26.20和13.86 kJ/mol,表明 Pt(1)与C(4)—O、Pt(1)与C(2)—C(3)存在较强的相互作用.M1和M2分别与H2结合形成中间体M3和M4.中间体M3中,Pt(2)—H(4)、C(4)——O键长分别为0.216 4和0.127 2 nm;中间体 M4中,Pt(2)—H(4)、C(2)—C(3)键长分别为0.165 9和0.142 7 nm.由表1能量数据可知,M1和M2与H2结合后体系能量进一步降低,表明中间体M3和M4易形成并能稳定存在.
在IA路径中,中间体M3经TS1→M5→TS5→M9过程得到产物 P3.过渡态 TS1中,五元环Pt(1)—Pt(2)—H(4)—C(4)—O的RCP电荷密度为0.020 5 a.u.;Pt(2)—H(4)和C(4)—O键长分别增加0.172 4和0.131 3 nm,BCP电荷密度分别降低0和0.328 1 a.u.;C(4)—H(4)键长为0.145 9 nm,BCP电荷密度为0.105 3 a.u..中间体M5中,C(4)—O和C(4)—H(4)键长分别为0.143 3和 0.110 2 nm,BCP电荷密度分别为0.250 6和0.273 1 a.u..以上构型数据表明,在M3→TS1→M5过程中,Pt(2)—H(4)键逐步断裂,C(4)—H(4)键逐步形成.过渡态TS5中,Pt(2)—H(5)、O—H(5)和C(4)—O键长分别为0.161 3、0.168 9和0.142 9 nm;中间体M9中,O—H(5)键长为0.097 4 nm,BCP电荷密度为0.339 7 a.u.,C(4)—O键长为 0.146 5 nm,BCP电荷密度为0.225 6 a.u..以上构型数据分析表明,在M5→TS5→M9过程中Pt(2)—H(5)键逐步断裂且O—H(5)键逐步形成,C(4)——O键加氢过程完成.根据表1能量数据可知,这一过程所需的活化能为98.42 kJ/ mol,是IA路径的速控步骤.最后,C——O键加氢产物与Pt18团簇解离形成P3.

在IB路径中,中间体M3经TS2→M6→TS6→M10过程形成产物 P3.过渡态 TS2中,Pt(2)—H(4)、O—H(4)和 C(4)—O键长分别增加至0.174 4、0.132 9和0.128 9 nm,相应的BCP电荷密度为0.105 5、0.120 7和0.363 4 a.u..中间体M6中,C(4)—O、O—H(4)键长分别为0.138 3和0.097 3 nm,BCP电荷密度分别为 0.278 0和0.340 2 a.u..上述构型参数说明,在M3→TS2→M6过程中,Pt(2)—H(4)键逐渐断裂同时 O—H(4)键逐步形成.过渡态TS6中,Pt(2)—H(5)键长增加至0.282 4 nm;C(4)—H(5)键长为0.166 2 nm,BCP电荷密度为0.081 0 a.u..中间体M10中,C(4)—H(5)、C(4)—O键长分别为0.109 4和0.143 5 nm,BCP电荷密度分别为 0.281 0和0.248 6 a.u..以上构型数据表明,在M6→TS6→M10过程中,Pt(2)—H(5)键逐步断裂,C(4)—H(5)键逐步形成.由表1能量数据表明,这一过程所需的活化能为128.79 kJ/mol,是IB路径的控制步骤.最后,C——O键加氢产物与Pt18团簇解离形成产物P3.
在IC路径中,M3→TS2→M6过程与IB路径中一致,而该路径中中间体M6经过渡态TS7形成中间体 M11,最后得到产物 P2.过渡态 TS7中,C(2)—H(5)、Pt(2)—H(5)和C(2)—C(3)键长分别为0.156 0、0.164 6和0.141 6 nm,相应的BCP电荷密度分别为0.082 7、0.125 8和0.300 8 a.u.;与中间体M6中C(3)—C(4)相比,键长缩短了0.003 2 nm,BCP电荷密度增加了0.019 7 a.u..中间体 M11中,C(2)—C(3)、C(2)—H(5)和C(3)—C(4)键长分别为 0.151 0、0.109 8和0.133 3 nm,相应的 BCP电荷密度为 0.250 9、0.270 7和0.347 4 a.u..以上构型数据表明,在M6→TS7→M11过程中 Pt(2)—H(5)键逐步断裂,C(2)—H(5)键逐步形成.由表1能量数据可知,该步骤的活化能为122.88 kJ/mol,是IC路径的速控步骤.中间体M11中,肉桂醛的1,4-加氢产物与Pt18团簇解离后得到产物P2.
在ID路径中,中间体M4经TS3→M7→TS8→M12过程形成产物 P2.过渡态 TS3中五元环Pt(1)—Pt(2)—H(4)—C(2)—C(3)的RCP电荷密度为0.018 4 a.u.;Pt(2)—H(4)、C(2)—H(4)和C(2)—C(3)键长分别为0.162 3、0.172 4和0.143 0 nm,相应的BCP电荷密度分别为0.129 9、0.057 6和0.294 2 a.u..中间体M7中,C(2)—H(4)、C(2)—C(3)键长分别为0.110 1和0.154 0 nm,BCP电荷密度分别为0.267 9和0.239 6 a.u.; Pt(2)—H(5)键长为0.156 2 nm.以上构型数据表明,在M4→TS3→M7过程中,Pt(2)—H(4)键逐渐断裂,C(2)—H(4)键逐渐形成.对表1能量数据分析可知,该过程的活化能为224.09 kJ/mol,是ID路径的速控步骤.过渡态TS8中,O—H(5)键长为0.164 7 nm,BCP电荷密度为 0.061 2 a.u.; Pt(2)—H(5)和C(4)—O键长分别增加至0.161 9和0.135 1 nm,C(3)—C(4)键长缩短至0.134 8 nm.中间体M12中,O—H(5)、C(3)—C(4)和C(4)—O键长分别为0.097 5、0.139 5和0.137 1 nm,相应的BCP电荷密度分别为0.337 7、0.315 2和0.283 3 a.u..以上构型数据表明,在M7→TS8→M12过程中Pt(2)—H(5)键逐步断裂,O—H(5)键逐步形成.最后,肉桂醛的1,4-加氢产物与Pt18团簇解离后得到产物P2.
在IE路径中,M4→TS3→M7过程与ID路径中一致,同样也是IE路径的速控步骤,中间体M7经过渡态TS9形成了中间体M13.过渡态TS9中,C(3)—H(5)键长为0.171 0 nm,BCP电荷密度为0.072 7 a.u.;Pt(2)—H(5)键长增加至0.158 6 nm.中间体M13中,C(3)—H(5)、C(2)—C(3)键长分别为0.180 3和0.155 8 nm,BCP电荷密度分别为0.271 5和0.226 4 a.u..以上构型数据说明,在M17→TS9→M13过程中Pt(2)—H(5)键逐步断裂,O—H(5)键逐步形成.最后C——C键加氢产物经过解离过程得到产物P1.
在IF路径中,中间体M4经TS4→M8→TS10→M14过程得到产物 P1.过渡态 TS4中五元环Pt(1)—Pt(2)—H(4)—C(3)—C(2)的RCP电荷密度为0.065 6 a.u.;Pt(2)—H(4)、C(3)—H(4)和C(2)—C(3)键长分别为0.200 6、0.155 3和0.145 7 nm,相应的BCP电荷密度分别为0.059 7、0.093 1和0.279 1 a.u..中间体M8中,C(3)—H(4)、C(2)—C(3)键长分别为0.110 6和0.152 9 nm,BCP电荷密度分别为0.261 6和0.243 6 a.u..以上构型数据表明,M4→TS4→M8过程中,Pt(2)—H(4)键逐渐断裂,C(3)—H(4)键逐渐形成.表1能量数据表明,这一过程是IF路径的速控步骤,活化能为128.13 kJ/mol.过渡态 TS10中,C(2)—H(5)键长为0.153 2 nm,BCP电荷密度为0.103 1 a.u.;C(2)—C(3)键长增加至0.160 0 nm.中间体M14中,C(2)—H(5)、C(2)—C(3)键长分别为0.109 6和0.155 1 nm,BCP电荷密度分别为0.273 1和0.232 0 a.u..以上构型参数说明,M8→TS10→M14过程中Pt(2)—H(5)键逐步断裂,C(2)—H(5)键逐步形成.最后,C——C加氢产物经过解离过程得到产物P1.
2.2 能量分析 从图3可以看出,肉桂醛中的C——O、C——C键分别与Pt18团簇活化络合后形成中间体M1和M2,能量分别降低43.14和80.20 kJ/ mol,再分别与H2结合形成中间体M3和M4,体系总能量进一步降低.中间体M3经IA、IB路径形成产物P3:IA路径的速控步骤为M5→TS5→M9,活化能为98.42 kJ/mol;IB路径的速控步骤为M6→TS6→M10,活化能为128.79 kJ/mol;因此,IA路径是形成产物P3的最优反应通道.由IC和ID路径得到产物P2:IC路径中速控步骤为M6→TS7→M11,活化能为122.88 kJ/mol;ID路径中速控步骤M4→TS3→M7的活化能为224.09 kJ/mol.比较可知,IC路径是形成产物P2的最优反应通道.经IE和IF路径可得到产物P1.IE路径中,M4→TS3→M7过程与ID路径中一致,这一过程也是IE路径的速控步骤.IF路径的速控步骤为M4→TS4→M8,活化能为128.13 kJ/mol.由此可知,IF路径是形成P1的最优反应通道.综合分析发现Pt18团簇催化肉桂醛选择性加氢反应中C——O键加氢所需的活化能最低.
3 结论
本文采用密度泛函理论(DFT)中的B3LYP方法,研究了Pt18团簇催化肉桂醛选择性加氢反应的机理.研究发现,Pt18团簇催化肉桂醛选择性加氢反应有6条不同的反应路径.反应过程很复杂,其中IA路径是形成产物肉桂醇(P3)的最优反应通道,速控步骤为M5→TS5→M9,活化能为98.42 kJ/ mol;IC路径是形成产物3-苯基丙烯醇(P2)的最优反应通道,速控步骤为M6→TS7→M11,活化能为122.88 kJ/mol;IF路径是形成3-苯基丙醛(P1)的最优反应通道,速控步骤为M4→TS4→M8,活化能为128.13 kJ/mol.综合分析发现Pt18团簇催化肉桂醛选择性加氢反应中C——O键加氢所需的活化能最低,有利于肉桂醇的生成.
[1]MAHATA N,PEREIRA M,FIGUEIREDO J L,et al.Selective hydrogenation of cinnamaldehyde to cinnamyl alcohol over mesoporous carbon supported Fe and Zn promoted Pt catalyst[J].Appl Catalysis:General,2008,A339:159-168.
[2]高雪霞,王春霞,刘忠文,等.炭材料负载Pt催化肉桂醛选择性加氢性能的研究[J].工业催化,2008,16(10):88-91.
[3]PRASHAR A K,MAYADEVI S,DEVI R N.Effect of particle size on selective hydrogenation of cinnamaldehyde by Pt encapsulated in mesoporous silica[J].Catalysis Commun,2012,28:42-46.
[4]张付利,李宇熙,李省.硼氢化钾还原肉桂醛合成肉桂醇的工艺研究[J].化学研究,2012,23(1):89-91.
[5]NERI G,ARRIGO I,CORIGLIANO F,et al.Support and solvent effects on the liquid-phase chemoselective hydrogenation of crotonaldehyde over Pt catalysts[J].Appl Catalysis:General,2010,A385:190-200.
[6]HANDJANI S,MARCEAU E,BLANCHARD J,et al.Influence of the support composition and acidity on the catalytic properties of mesoporous SBA-15,Al-SBA-15,and Al2O3-supported Pt catalysts for cinnamaldehyde hydrogenation[J].J Catalysis,2011,282:228-236.
[7]SHI J J,NIE R F,CHEN P,et al.Selective hydrogenation of cinnamaldehyde over reduced grapheme oxide supported Pt catalyst[J].Catalysis Commun,2013,41:101-105.
[8]GUTI RREZ V,NADOR F,RADIVOY G,et al.Highly selective copper nanoparticles for the hydrogenation of α,β-unsaturated aldehydes in liquid phase[J].Appl Catalysis:General,2013,A464/465:109-115.
[9]毛华平,杨兰蓉,朱正和.Yn(n=2~8)小团簇的结构与稳定性研究[J].四川师范大学学报(自然科学版),2006,29(2): 228-231.
[10]张萍,杨继先,汪勇.PdnAu(n=1~5)团簇的结构性质研究[J].四川师范大学学报(自然科学版),2008,31(2):328-331.
[11]肖培,刘以良,姜明.密度泛函理论对AlnTi2(n=2~11)团簇结构和稳定性的研究[J].四川师范大学学报(自然科学版),2014,37(3):369-374.
[12]KALITA B,DEKA R C.DFT study of CO adsorption on neutral and charged Pdn(n=1~7)clusters[J].Europ Phys J D,2009,53:51-58.
[13]CHOU J P,CHEN H Y T,HSING C R,et al.13-atom metallic clusters studied by density functional theory:dependence on exchange-correlation approximations and pseudopotentials[J].Phys Rev,2009,B80:165412(1-10).
[14]BALBUENA P E,CALVO S R,LAMAS E J,et al.Adsorption and dissociation of H2O2on Pt and Pt-alloy clusters and surfaces[J].J Phys Chem,2006,B110:17452-17459.
[15]LAREF S,DELBECQ F,LOFFREDA D.Theoretical elucidation of the selectivity changes for the hydrogenation of unsaturated aldehydes on Pt(111)[J].J Catalysis,2009,265:35-42.
[16]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations:potentials for the transition metal atoms Sc to Hg[J].J Chem Phys,1985,82(1):270-283.
[17]REED A J,CURTISS L W,WEINHOLD F.Intermolecular interactions form a natural bond orbital,donor-acceptor viewpoint[J].Chem Rev,1988,88(6):899-926.
[18]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09[S].Rev A.2nd.Wallingford CT:Gaussian Inc,2009.
[19]BADER R W F.Atoms in Molecules:a Quantum Theory[M].Oxford:Oxford University Press,1990.
[20]BAEZA B B,RAMOS I R,RUIZ A G.Influence of Mg and Ce addition to ruthenium based catalysts used in the selective hydrogenation of α,β-unsaturated aldehydes[J].Applied Catalysis:General,2001,A205(1/2):227-237.
[21]NHUT J M,VIEIRA R,PESANT L,et al.Synthesis and catalytic uses of carbon and silicon carbide nanostructures[J].Catalysis Today,2002,76(1):11-32.
[22]邹鸣,牟新东,颜宁,等.离子液体中离子型共聚高分子保护的铂纳米粒子催化剂催化肉桂醛选择性加氢[J].催化学报,2007,28(5):389-391.
[23]余建雁,宋绍飞,叶素芳,等.可循环磁性Pt/Fe3O4-MCNT催化剂上的肉桂醛选择性加氢反应[J].无机化学学报,2014,30(2):271-276.
[24]孙芳芳,张林,马骏,等.肉桂醛选择性加氢产物的互变异构理论研究[J].四川师范大学学报(自然科学版),2014,38(5):697-702.
Investigation on the Mechanism for Cinnamic Aldehyde Selective Hydrogenation Reaction Catalyzed by Pt18Cluster
WEI Wei,WANG Wei,LI Laicai
(College of Chemistry and Material Science,Sichuan Normal University,Chengdu 610066,Sichuan)
The reaction mechanism on cinnamic aldehyde selective hydrogenation reaction catalyzed by Pt18cluster was studied by the density functional theory(DFT).The geometries and the frequencies of reactants,intermediates,transition states and products have been calculated at the B3LYP/6-31+G(d,p)level.Transition states have been confirmed by the corresponding vibration analysis and intrinsic reactions coordinate(IRC).The results showed that there were six different reaction paths in cinnamic aldehyde selective hydrogenation reaction catalyzed by Pt18cluster.The activation energies of rate-determining steps in which cinnamic aldehyde selective hydrogenation reaction catalyzed by Pt18cluster to form C——O adduct was 98.42 kJ/mol,and C——C adduct was 122.88 kJ/mol.The final result of the theory study showed that the activation energies of C——O adduct was lower in cinnamic aldehyde selective hydrogenation reaction catalyzed by Pt18cluster.
density functional theory;Pt cluster;cinnamic aldehyde;selective hydrogenation reaction
O561.4
A
1001-8395(2016)05-0711-08
10.3969/j.issn.1001-8395.2016.05.017
(编辑 李德华)
2015-10-21
四川省教育厅自然科学重点基金(13ZA0150)和四川省科技厅应用基础基金(2014JY0099)
*通信作者简介:李来才(1966—),男,教授,主要从事应用量子化学的研究,E-mail:lilcmail@163.com
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
汕头大学学报(自然科学版)(2020年4期)2020-12-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
广西林业科学(2016年1期)2016-03-20
广西林业科学(2016年1期)2016-03-20
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
股市动态分析(2015年12期)2015-09-10
物理化学学报(2015年5期)2015-02-28
天然产物研究与开发(2014年8期)2014-04-27
