
Increasing isobutanol yield by double-gene deletion of PDC6 and LPD1 in Saccharomyces cerevisiae☆
2016-06-01 03:01AiliZhangYangLiYuhanGaoHongxingJin
Aili Zhang,Yang Li,Yuhan Gao,Hongxing Jin
School of Chemical Engineering and Technology,Hebei University of Technology,Tianjin,300130,China
1.Introduction
With fossil fuel resource decreasing and serious pollution caused by fossil fuel combustion, biofuel energy attracts more and more attentions[1-4].Bioethanol has been recognized as a useful biofuel.Compared to ethanol,isobutanol has more advantages,such as lower hygroscopicity,higher energy density and higher-octane value[2,3].It has been reported that isobutanol can be synthesized by engineered microbes[4],such asEscherichia coli[5],Clostridium cellulolyticum[6],Bacillus subtilis[7],Corynebacterium glutamicum[8],Synechocystissp.Strain PCC 6803[9],Klebsiella pneumonia[10],Zymomonas mobilis[11],andSaccharomyces cerevisiae[12].It is known thatS.cerevisiaeproduces alcohols naturally,such as ethanol and isobutanol[13]and it has more advantages in isobutanol production than other microorganisms[14,15]due to its robustness and tolerance to low pH.In this study,S.cerevisiaeis used to produce isobutanol.
As illustrated in Fig.1,isobutanol biosynthesis is based on the valine metabolic pathway inS.cerevisiae.Pyruvate is converted to 2-ketoisovalerate catalyzed by acetolactate synthase (Ilv2p),acetohydroxyacid reductoisomerase(Ilv5p),and dihydroxyacid dehydratase(Ilv3p)[12].The bidirectional reaction between 2-ketoisovalerate and valine is catalyzed by branched-chain amino-acid aminotransferases(BAT1andBAT2,present in mitochondrial matrix and cytosol,respectively).Finally,2-ketoisovalerate is converted to isobutanol by pyruvate decarboxylases(encoded byARO10)and alcohol dehydrogenases(ADH).
Severalmethods have been used for higher isobutanol production inS.cerevisiae,such as overexpression of genes in valine biosynthesis pathway(ILV2,ILV5,ILV3,BAT2,KDCsandADHs)[12,16-18].Expressing valine biosynthesis genes in cytoplasm or expressing pyruvate decarboxylase in mitochondria could enhance isobutanol production[19-21].Higher isobutanol production has been achieved by deletingALD6andBAT1to eliminate competing pathways and increasing transcription of endogenous genes in the valine and leucine biosynthetic pathway by expressingLeu3△601,which is a constitutively active form ofLeu3transcriptional activator[22].Isobutanol production could be increased by deletingLPD1encoding pyruvate dehydrogenase,which competes for pyruvate available,and by overexpressing NADPH-generating malice enzyme to resolve cofactor imbalance[23].Singlegene deletion ofPDC1,PDC5orPDC6could increase isobutanol production[17,18],but integration effects of double-gene deletion ofLPD1andPDC6in strains carrying overexpressedBAT2andILV2have not well understood.

Fig.1.Isobutanol and ethanol biosynthesis pathways in S.cerevisiae.
In this study,integration effects of double-gene deletion ofLPD1andPDC6on isobutanol production are investigated,whenLPD1andPDC6are deleted in strains overex pressingILV2andBAT2.Fermentation characters(such as growth rates,glucose consumption rates,biomass concentrations at the end of fermentation,ethanol formation,glycerol formation and acetic acid titer)of these engineered strains are also examined.The results of this study willlay the foundation forfuture development of the engineered strain for production of isobutanol.
2.Materials and Methods
2.1.Strains and mediums
YeastS.cerevisiaeW303-1A[24]was used as a controlstrain.The relevant genotypes for strains in this study were listed in Table 1.Luria-Bertani medium(1%tryptone,0.5%yeast extract and 1%NaCl)with ampicillin was used for plasmid preparation fromE.coliTOP 10'.Yeast extract peptone dextrose medium(2%peptone,1%yeast extract,2%glucose)was used to rout inely maintain and propagate yeaststrains.Synthetic complete(SC)media(0.67%bacto yeast nitrogen base without amino acids supplemented with appropriate amino acids and 2%D-glucose)were used for selection of transformants.5-Fluoroorotic acid hydrate(5-FOA)medium(1.5%agar,0.67%bacto yeast nitrogen base without amino acids,1.7 g⋅L-1dropout power without amino acids,2%D-glucose,50 mg⋅L-1adenine,150 mg⋅L-1histidine,100 mg⋅L-1tryptophan,200 mg⋅L-1leucine,0.2%5- fluoroorotic acid hydrate)was used to selectura3-cells.

Table 1Strains used in this study
2.2.Constructions of engineered strains
2.2.1.Over-expression of BAT2 gene
Primers and plasmids used in this study were described in Tables 2 and 3,respectively.
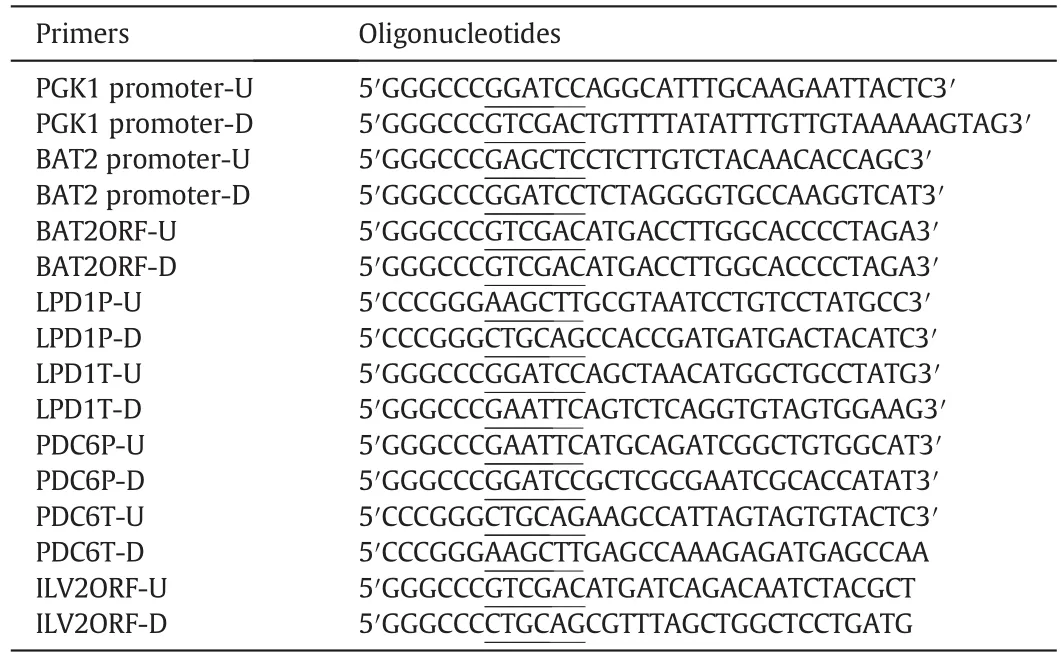
Table 2Primers used in this study
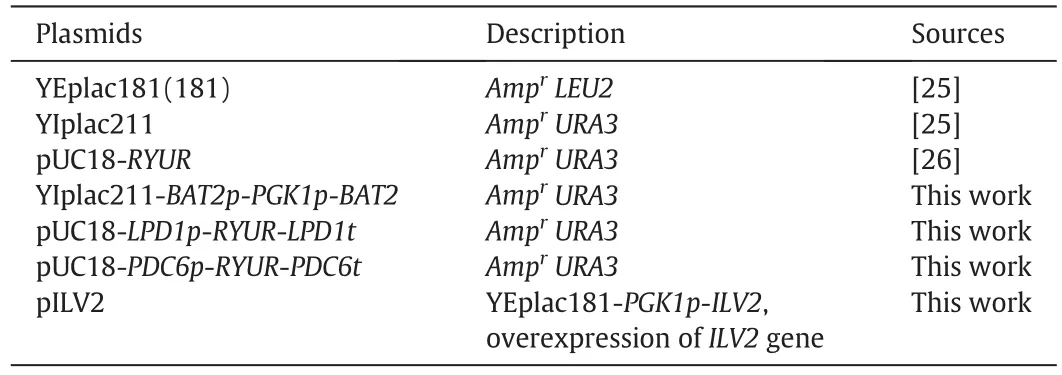
Table 3Plasmids used in this study
To overexpressBAT2gene,an integration plasmid with bothPGK1promoter and coding sequence ofBAT2gene was constructed.First,the promoter sequence ofBAT2was cloned by PCR with primers BAT2promoter-U and BAT2promoter-D and inserted into theSacI andBamHI sites of plasmid YIplac211,resulting in plasmid YIplac211-BAT2p.Then part of theBAT2ORF sequence was inserted into plasmid YIplac211-BAT2p by cloningSalI andPstI-treated PCR products(amplified by primers BAT2ORF-U and BAT2ORF-D),resulting in plasmid YIplac211-BAT2p-BAT2.The plasmid YIplac211-BAT2p-BAT2 was digested withSalI andBamHI and ligated toSalI andBamHI-treated PCR fragment containingPGK1promoter(amplified by primers PGK1promoter-U(BamHI)and PGK1promoter-D(SalI)).The resulting plasmid was denoted as YIplac211-BAT2p-PGK1p-BAT2.After being digested bySpeІ,YIplac211-BAT2p-PGK1p-BAT2 was used to transform into W303-1A using the lithium acetate method.Cells were plated onto SC minus uracilmedium plates.Correctinsertion of the plasmid into theBAT2locus on chromosome was verified by PCR with primers BAT2promoter-U and BAT2ORF-D.Finally,to loop-out ofURA3marker gene by homologous recombination of the two directBAT2promotersequences,correct transformations were cultivated on 5-FOA plates,and mutants were verified by PCR with primers BAT2promoter-U and BAT2ORF-D.Correct engineered strains containingPGK1p-BAT2in place of endogenousBAT2was denoted as HZAL-2(MATaPGKp-BAT2)(Table 1).
2.2.2.Over-expression of ILV2 gene
To overexpressILV2,plasmid YEplac181-PGK1p-ILV2was constructed by inserting the constitutive promoter ofPGK1in front ofILV2'open reading frame into YEplac181.PGK1pwas amplified by PCR using primers PGK1promoter-U(containingBamHI enzyme site)and PGK1promoter-D(containingSalI enzyme site).ILV2was amplified by PCR using primers ILV2ORF-U(containingSalI enzyme site)and ILV2ORF-D(containingPstI enzyme site).Plasmid YEplac181-PGK1p-ILV2was also introduced into mutants using the lithium acetate method and the trans for mants were selected on SC minus leucine medium.W303-1A transformed with the empty plasmid was used as control strain.
2.2.3.Knockout of PDC6 gene
Plasmid pUC18-PDC6p-RYUR-PDC6twas constructed for disruptingPDC6by inserting partof5′-end ofPDC6promotersequence(containingEcoRI enzyme site andBamHI enzyme site),a short fragment(denoted as R)(containingBamHI enzyme site andXbaI enzyme site)ofB.subtilischromosomal DNA,URA3gene(containingXbaI enzyme site andSalI enzyme site),an inverted fragment R(containingSalI enzyme site andPstI enzyme site),and a segment of 3′-end region ofPDC6(containingPstI enzyme site andHindIII enzyme site)into pUC18.Plasmid pUC18-PDC6p-RYUR-PDC6tdigested withEcoRІandHindШ,and the cassette containingPDC6p-RYUR-PDC6twas transformed into HZAL-2 using the lithium acetate method.Correct transformants were selected by PCR.Finally,RYUR fragment includingURA3marker was pop out by homologous recombination of the two direct repeat fragments R on 5-FOA media.Correct engineered strain was denoted as HZAL-7(Table 1).
2.2.4.Knockout of LPD1 gene
Plasmid pUC18-LPD1p-RYUR-LPD1twas constructed for disruptingLPD1,by inserting partof5′-end ofLPD1promotersequence(containingHindIII enzyme site andPstI enzyme site),a short fragment(denoted as R)(containingPstI enzyme site andSalI enzyme site)ofB.subtilischromosomal DNA,URA3gene(containingSalI enzyme site andXbaI enzyme site),an inverted fragment R(containingXbaI enzyme site andBamHI enzyme site),and a segment of 3′-end region ofLPD1(containingBamHI enzyme site andEcoRI enzyme site)into pUC18.Plasmid pUC18-LPD1p-RYUR-LPD1tdigested withEcoRІ andHindШ,and the cassette containingLPD1p-RYUR-LPD1twas transformed into HZAL-2 using the lithium acetate method.Correct transformants were selected by PCR.And correct engineered strain was denoted as HZAL-12(Table 1).Meanwhile,the cassette containingLPD1p-RYUR-LPD1twas transformed into HZAL-7 using the lithium acetate method,and correct engineered strain was denoted as HZAL-11.
2.3.Fermentations and measurements of fermentation products
Synthetic complete minus leucine(SC-leucine)media(0.67%bacto yeast nitrogen base without amino acids supplemented with appropriate amino acids and 40 g⋅L-1glucose)were used for fermentations.Fermentations were performed in triplicate and representative cultivations were shown.
Middle log phase cells in the pre-cultivation were harvested,and then inoculated into 100 ml of SC-leucine medium with 40 g⋅L-1glucose.Fermentations were performed at 30 °C with 100 r·min-1agitation to create micrometric condition.The initial OD600of the fermentations were adjusted to 0.2.Samples were collected during fermentation.Growth curves were measured based on OD600using a UV-721G spectrophotometer.Glucose concentrations were determined using DNS methods.Concentrations of ethanol,glycerol,and acetic acid were determined by a high-performance liquid chromatography system(Agilent Technologies 1260 Series)equipped with a Carbomix H-NP column and a refractive index(RI)detector.The column was eluted with 2.5 mmol·L-1H2SO4ata flow rate of0.6 ml⋅min-1at55 °C and RI detector was kept at 35°C.Isobutanol concentration was quantified by a gas chromatography system(Bruker 456-GC)equipped with a HP-INNOWAX column(length of 60 m,inner diameter of 0.32 mm).The column temperature wascontrolled at80°C for10 min.The injector and detector temperatures were maintained at 200 °C and 300 °C,respectively.
2.4.Determination of cell growth and dry weight
The growths ofyeastcells were measured by reading the absorbance of the culture at 600 nm.Samples were centrifuged at5000 g for 10 min and washed twice with water.Subsequently the pellet was dried at 100°C for 24 h and weighed.
3.Results
To investigate integration effects of overexpression ofILV2/BAT2and double-gene deletion ofLPD1andPDC6on isobutanol production,engineered strains HZAL-2 pILV2,HZAL-7 pILV2,HZAL-11 pILV2 and HZAL-12 pILV2 were constructed from W303-1A.Their fermentation characters were examined and shown in Fig.2 and Table 4.The growth rate of control strain(W303-1A(181))was higher than that of these engineered strains,while glucose consumption rate of the controlstrain was faster.Overexpression ofILV2andBAT2gave lower biomass concentration(Table 4),while additional deletion ofLPD1further reduced biomass concentration.
As shown in Fig.3A,the control strain(W303-1A(181))produced 7.4 mg⋅L-1isobutanolat30 h in SC-leucine mediumwith 40 g⋅L-1glucose,overexpression ofBAT2andILV2(HZAL-2 pILV2)resulted in a higher isobutanol production(33 mg⋅L-1)at 30 h,additional deletion ofPDC6in strains carrying overexpressedBAT2andILV2(HZAL-7 pILV2)generated 49 mg⋅L-1isobutanol at 30 h,while HZAL-2 pILV2 and HZAL-7 pILV2 generated similar isobutanol titers(about 18 mg⋅L-1)at 48 h,indicating that the appropriate time for isobutanol fermentation with engineered strains HZAL-2 pILV2 and HZAL-7 pILV2 are 30 h.We also found that additional deletion ofLPD1in strains carrying overexpressedBAT2andILV2(HZAL-12 pILV2)generated 39.5 mg⋅L-1isobutanol at 48 h,while deletion both ofLPD1andPDC6in strains carrying overexpressedBAT2andILV2(HZAL-11 pILV2)increased isobutanol titer(59.9 mg⋅L-1)by 8.1-fold compared with control strain(W303-1A(181)).Thus deletion of bothLPD1andPDC6in strains overexpressing ofBAT2andILV2could increase isobutanol titer dramatically than single-gene deletion ofLPD1orPDC6.

Fig.2.Growth(A)and glucose consumption(B)ofstrains carrying plasmids 181 or pILV2.W303-1A 181;HZAL-2 pILV2;▲HZAL-7 pILV2;HZAL-11 pILV2;HZAL-12 pILV2.
Ethanol is one of the main products in fermentations ofS.cerevisiae.As shown in Fig.3B,the maximum ethanol titer was 6.99 g⋅L-1in control strain(W303-1A(181)).The maximum ethanol titers of HZAL-2 pILV2,HZAL-7 pILV2,HZAL-11 pILV2,and HZAL-12 pILV2 were 2.64,4.45,5.19,and 4.58 g⋅L-1,respectively.Thus the maximum ethanol titers of these engineered strains decreased markedly than thatof control strain.Their lower ethanol titers are perhaps due to the lower growth rates(Fig.2A)and lower glucose consumption rates(Fig.2B).
As shown in Fig.3C and D,the maximum glycerol titer and the maximum acetic acid titer of control strain(W303-1A(181))were 0.515 and 0.103 g⋅L-1,respectively.Acetic acid titers of engineered strains HZAL-2 pILV2,HZAL-7 pILV2,HZAL-11 pILV2,and HZAL-12 pILV2 were higher than that of the control strain,and they produced 0.14,0.29,0.15,and 0.245 g⋅L-1acetic acid,respectively,at 24 h.Our data suggest that glycerol titers of these four engineered strains do not change significantly as that of the control strain(Fig.3C).Thus the deletion ofPDC6orLPD1does not have significant effects on glycerol biosynthesis.
4.Discussion
Our results showed that control strain(W303-1A(181))produced about 7.4 mg⋅L-1isobutanol at 30 h in SC-leucine medium with 40 g⋅L-1initial glucose.Overexpression ofBAT2andILV2(HZAL-2 pILV2)could increase isobutanol titer by 4.4-fold compared with the control strain.Additional deletion ofPDC6in strains carrying overexpressedBAT2andILV2(HZAL-7 pILV2)produced about 1.5-fold increase in isobutanol titer than engineered strain HZAL-2 pILV2.It is well known that pyruvate decarboxylases(PDCs)catalyzed both 2-ketoisovalerate converting to isobutanol and pyruvate converting to acetaldehyde.Higher isobutanol titer ofpdc6Δ strains carryingBAT2andILV2indicated that Pdc6p might have higher af finities to pyruvate than to 2-ketoisovalerate.Our results also indicated that additional deletion ofLPD1in strains carrying overexpressedBAT2andILV2(HZAL-12 pILV2)generated a 5.3-fold higher isobutanol titer compared with the control strain at 48 h.The deletion of bothLPD1andPDC6mutant in strains carrying overexpressedBAT2andILV2(HZAL-11 pILV2)increased isobutanol titer by 8.1-fold compared with the control strain and by 1.5-fold compared with engineered strain HZAL-12 pILV2 at 48 h.Thus the deletion of bothLPD1andPDC6in strains overexpressing ofBAT2andILV2could improve isobutanol titer dramatically than single-gene deletion ofLPD1orPDC6.These data also indicated that affinities of pyruvate decarboxylases to 2-ketoisovalerate might be one of the main factors affecting isobutanol titer.Introducing pyruvate decarboxylases with higher af finities to 2-ketoisovalerate(such as Aro10)inpdc6Δlpd1Δ strains carrying overexpressedBAT2andILV2might further increase isobutanol titer.In this study,onlyILV2(encoding acetolactate synthase that catalyzes the first step in valine synthesis)was overexpressed.Isobutanolmightfurtherimproved by simultaneously overexpressingILV5andIVL3.
Ethanol titers of engineered strains(HZAL-2 pILV2,HZAL-7 pILV2,HZAL-12 pILV2,HZAL-11 pILV2)decrease more markedly than control strain(W303-1A(181)).Lower ethanol titers of these strains are perhaps due to lower growth rates and lower glucose consumption rates.Meanwhile,higher isobutanol titer and lower ethanol titer inpdc6Δ mutant carrying overexpressedBAT2/ILV2indicate that pyruvate decarboxylases encoded byPDC6may have higher affinity to pyruvate than to 2-ketoisovalerate.
Our data also show thatacetic acid titers ofengineered strains HZAL-2 pILV2,HZAL-7 pILV2,HZAL-12 pILV2 and HZAL-11 pILV2 are higher than that of the control strain.This suggests that,to balance the imbalance of NAD(P)H/NAD(P)+produced via valine biosynthetic pathway,higher acteic acid accompanied by more NADPH/NADH are synthesized in these engineered strains.In addition,overex pressing NADPH/NADH-generating pathwaysto resolve cofactorimbalance may furtherincrease isobutanol titer.It has been reported that dihydroxyacetone converts to glycerol catalyzed by NAD-dependent glycerol-3-phosphate dehydrogenase(encoded byGPD1/GPD2)and glycerol-3-phosphatase(encodedbyGPP1/GPP2)[26].The first reaction catalyzed by NAD-dependent glycerol-3-phosphate dehydrogenase is the restriction step,which produced NAD+.Deletion ofGPD1orGPD2or both will perhaps increase isobutanol titer due to eliminating glycerol biosynthesis that is competing carbon sources with isobutanol biosynthesis and decreasing NAD+biosynthesis.However,we have found that glycerol titers of engineered strains HZAL-2 pILV2,HZAL-7 pILV2,HZAL-12 pILV2 and HZAL-11 pILV2 do not change significantly as that of the control strain.This indicates that eliminating glycerolbiosynthesis may not affectisobutanol synthesis markedly.Other pathways generating more NADPH/NADH to resolve cofactor imbalance may be found.
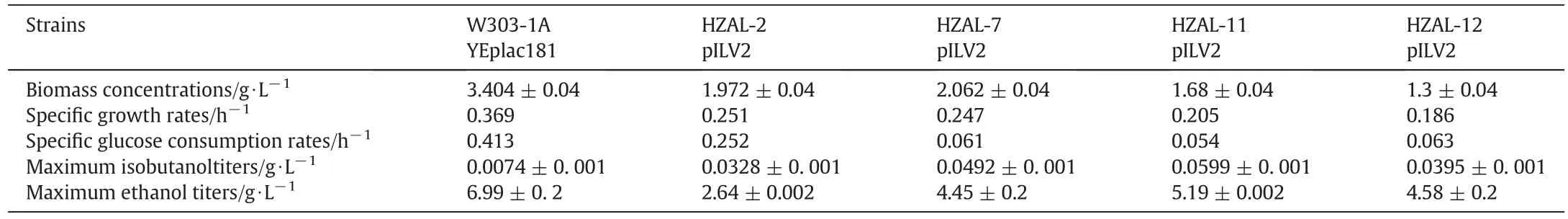
Table 4Comparison of growth and compound titers of engineered strains in batch fermentations

Fig.3.Changes in measured parameters of isobutanol titers(A),productions of ethanol(B),formations of glycerol(C),and formation of acetic acid(D)during batch fermentations of engineered strains(W303-1 A,HZAL-2,HZAL-7,HZAL-11,HZAL-12)carrying plasmids 181 or pILV2 with 40 g⋅L-1 glucose as carbon source,with the means and standard deviations for triplet experiments.
5.Conclusions
In conclusion,double-gene deletion ofbothLPD1andPDC6in strains overexpressing ofBAT2andILV2could increase isobutanoltiter dramatically than single-gene deletion ofLPD1orPDC6.Thus it is effective to improve isobutanoltiterby enhancing the activity ofvaline biosynthetic pathway(via overexpression ofILV2andBAT2)combined with eliminating competing pathways including ethanol biosynthesis(via deletion ofPDC6)and acetyl-CoA biosynthesis(via deletion ofLPD1).
[1]K.Kuroda,M.Ueda,Cellular and molecular engineering of yeastSaccharomyces cerevisiaefor advanced biobutanol production,FEMS Microbiol.Lett.363(3)(Feb 2016).
[2]C.Weber,A.Farwick,F.Benisch,et al.,Trends and challenges in the microbial production of lignocellulosic bioalcohol fuels,Appl.Microbiol.Biotechnol.87(4)(2010)1303-1315.
[3]H.Sakuragi,K.Kuroda,M.Ueda,Molecular breeding of advanced microorganisms for biofuel production,J.Biomed.Biotechnol.1(17)(2011)1-11.
[4]S.Atsumi,T.Hanai,J.C.Liao,Non-fermentative pathways for synthesis of branchedchain higher alcohols as biofuels,Nature451(7174)(2008)86-89.
[5]B.Blombach,B.J.Eikmanns,Current knowledge on isobutanol production withEscherichia coli,Bacillus subtilisandCorynebacterium glutamicum,Bioeng.Bugs2(6)(2011)346-350.
[6]W.Higashide,Y.Li,Y.Yang,J.C.Liao,Metabolic engineering ofClostridium cellulolyticumfor production of isobutanol from cellulose,Appl.Environ.Microbiol.77(8)(2011)2727-2733.
[7]S.Li,J.Wen,X.Jia,EngineeringBacillus subtilisfor isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression,Appl.Microbiol.Biotechnol.91(3)(2011)577-589.
[8]K.M.Smith,K.M.Cho,J.C.Liao,EngineeringCorynebacterium glutamicumfor isobutanol production,Appl.Microbiol.Biotechnol.87(3)(2010)1045-1055.
[9]A.M.Varman,Y.Xiao,P.HB,et al.,Metabolic engineering ofSynechocystis sp.strain PCC 6803 for isobutanol production,Appl.Environ.Microbiol.79(3)(2013)908-914.
[10]B.-R.Oh,S.-Y.Heo,S.-M.Lee,et al.,Erratum to production of isobutanol from crude glycerol by a genetically engineeredKlebsiella pneumoniaestrain,Biotechnol.Lett.10(25)(2013)1-6.
[11]M.X.He,B.Wu,H.Qin,et al.,Zymomonas mobilis:A novel platform for future biore fineries,Biotechnol.Biofuels7(101)(2014)1-15.
[12]X.Chen,K.F.Nielsen,I.Borodina,et al.,Increased isobutanol production inSaccharomyces cerevisiaeby overexpression of genes in valine metabolism,Biotechnol.Biofuels4(21)(2011)1-12.
[13]K.K.Hong,Nielsen,Metabolic engineering ofSaccharomyces cerevisiae:A key cell factory platform for future biore fineries,Cell.Mol.Life Sci.69(16)(2012)2671-2690.
[14]E.P.Knoshaug,M.Zhang,Butanol tolerance in a selection of microorganisms,Appl.Biochem.Biotechnol.153(2009)13-20.
[15]P.Fatehi,Recent advancements in various steps of ethanol,butanol,and isobutanol productions from woody materials,Biotechnol.Prog.29(2)(2013)297-310.
[16]T.Kondo,H.Tezuka,J.Ishii,et al.,Genetic engineering to enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from glucose bySaccharomyces cerevisiae,J.Biotechnol.159(1-2)(2012)32-37.
[17]W.-H.Lee,S.-O.Seo,Y.-H.Bae,et al.,Isobutanol production in engineeredSaccharomyces cerevisiaeby overexpression of 2-ketoisovalerate decarboxylase and valine biosynthetic enzymes,Bioprocess Biosyst.Eng.35(9)(2012)1467-1475.
[18]E.Ofuonye,K.Kutin,D.T.Stuart,EngineeringSaccharomyces cerevisiaefermentative pathways for the production of isobutanol,Biofuels4(2)(2013)185-201.
[19]D.Brat,E.Boles,Isobutanol production from D-xylose by recombinantSaccharomyces cerevisiae,FEMS Yeast Res.13(2013)241-244.
[20]D.Brat,C.Weber,W.Lorenzen,et al.,Cytosolic re-localization and optimization of valine synthesis and catabolism enables increased isobutanol production with the yeastSaccharomyces cerevisiae,Biotechnol.Biofuels5(65)(2012)1-16.
[21]J.L.Avalos,G.R.Fink,G.Stephanopoulos,Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols,Nat.Biotechnol.31(2013)335-341.
[22]K.Ida,J.Ishii,F.Matsuda,et al.,Eliminating the isoleucine biosynthetic pathway to reduce competitive carbon out flow during isobutanol production bySaccharomyces cerevisiae,Microb.Cell Factories14(2015)62-70.
[23]F.Matsuda,J.Ishii,T.Kondo,et al.,Increased isobutanol production inSaccharomy cescerevisiaeby eliminating competing pathways and resolving cofactor imbalance,Microb.Cell Factories12(2013)119-129.
[24]B.J.Thomas,R.Rothstein,Elevated recombination rates in transcriptionally active DNA,Cell56(4)(1989)619-630.
[25]R.D.Gietz,A.Sugino,New yeast-Escherichia colishuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites,Gene74(2)(1988)527-534.
[26]A.L.Zhang,X.Chen,Improve ethanoltiterthrough minimizing glyceroltiter in ethanol fermentation ofSaccharomyces cerevisiae,Chin.J.Chem.Eng.16(4)(2008)620-625.
 Chinese Journal of Chemical Engineering2016年8期
Chinese Journal of Chemical Engineering2016年8期
- Chinese Journal of Chemical Engineering的其它文章
- Computational chemical engineering - Towards thorough understanding and precise application☆
- A review of control loop monitoring and diagnosis:Prospects of controller maintenance in big data era☆
- Experimental and numerical investigations of scale-up effects on the hydrodynamics of slurry bubble columns☆
- The heat transfer optimization of conical fin by shape modification
- The steady-state and dynamic simulation of cascade distillation system for the production of oxygen-18 isotope from water☆
- Experimental mass transfer coefficients in a pilot plant multistage column extractor