
Cloning, expression and characterization of a feruloyl esterase C from Penicillium chrysogenum
2016-05-25 09:58LVShan-shanLIGui-
科技视界 2016年12期
关键词:责任编辑
LV+Shan-shan LI+Gui-long YANG+Shao-long

【Abstract】Objective: To clone feruloyl esterase gene C from Penicillium chrysogenum and characterize the general properties of the enzyme. Methods: The feruloyl esterase C gene was amplified by PCR based on the Penicillium chrysogenum feruloyl esterase C gene sequence and cloned into the expression vector pPIC9K, resulting the recombinant plasmid pPIC9K-PcfaeC. The recombiant plasmid was linerized and transformed into P. pastoris by electroporation. The transformants was screened based on the transparent zone technology. The screened transformants was then induced by methanol. the enzymatic properties of the protein were then measured. Results: SDS-PAGE analysis showed that the molecular mass of the enzyme was about 30 kD. The length of the gene was 762 bp. It comprised one open reading framwork(ORF) and annotated to encode 249 amino acid. The optimal temperature and pH was found to be 40℃ and 6, respectively. Moreover, the recombinant enzyme was stable at 40-50℃ and pH 5-7. Conclusion: The enzyme successfully expressed in P. pastoris could laid theoretical foundation in food, fodder and paper making industry.
【Key words】Penicillium chrysogenum; Clone; Expression; Feruloyl esterase; Enzyme activity
Feruloyl esterase (FAE), also known as cinnamoyl esterases, cinnamoyl ester hydrolases and chlorogenate esterases, which could hydrolases feruloylated oligosaccharides, ferulic acid methylester and polysaccharide ferulic acid esters to release the antioxidative ferulic acid[1,2]. Feruloyl esterase could also in synergy with hemicellulase to promote the cellulose degradation thoroughly.It plays important roles in bioresource degradation and has its potential application in fodder, food and paper industry[3]. Feruloyl esterase were originally classified into two groups based on their induction and substrate specificity[4]. While with the publication of gene sequence and protein sequence and further based on the sequence homology and biochemical analysis, four putative types (A-D) were proposed[5]. The type-C TsFaeC could perform a transesterification reaction to yield feruloyl arabinose from MFA and L-arabinose[6] and StFaeC showed a strong preference for short chain alkyl ferulates, such as MFA[7].
Feruloyl esterase were found in many kinds of bacterial and fungi. It was reported that Lactobacillus sp[8], Streptomyces sp, Clostridium thermocellum[9], Neurospora crassa[10], A. nidulans, Aspergillus niger[11,12], A. flavus[13], and A. usamil[14], could produce feruloyl esterases. Generally, feruloyl esterase activity of parental strain is very low. Some microorganism even though possess the feruloyl esterase genes, but could not produce the feruloyl esterase such as P. chrysogenum. Thus the industrial application of the feruloyl esterase was limited. Ordinally, gene engineering method was often used to express genes. To date, the most favorable gene expression system included E.coli, filamentous fungi and Pichia Pastoris. Among these P. Pastoris expression system was widely applied in the production of non-starch polysaccharide hydrolytic enzymes [15]. In this study, we cloned the feruloyl esterase C gene from the strain P. chrysogenum HA4089 preserved in our laboratory by the PCR. It was successfully expressed in P. Pastoris GS115 and we further characterize the general properties of the recombinant enzyme.
1 Materials and Method
1.1 Materials and regents
P. chrysogenum HA4089 and Escherichia coli DH5α preserved in our laboratory; P. Pastoris GS115 and pPIC9K vector was purchased from Invitrogen; E. coli DH5α and P. pastoris GS115s compenent cells were made according to Molecular cloning manual[16]; Taq DNA polymerase, pfu DNA polymerase and pMD18-T was purchased from Bioteke; Plasmid Extraction Kit and Gel Extraction purification Kit was purchased from OMEGA; T4 DNA Ligase and various restriction enzymes were purchased from Takara; All primer synthesis and sequencing were accomplished by sangon. Ethyl ferulate was purchased from Shanghai hengyuan biological technology company; unless stated, all other regents were analytical grade.
1.2 Medium
PDA medium was used to activate and culture P. chrysogenum HA4089; LB medium and YPD medium were used to cultivate Escherichia Coli and P. Pastoris GS115; MD and MM medium was used to screen yeast transformants; BMMY and BMGY medium were used to induce the protein expression. The component of YPD, MD, MM, BMMY and BMGY medium was prepared according to P. pastoris operation brochure provided by Invitrogen (www. invitrogen.com).
1.3 cDNA cloning from the strain P. chrysogenum HA4089
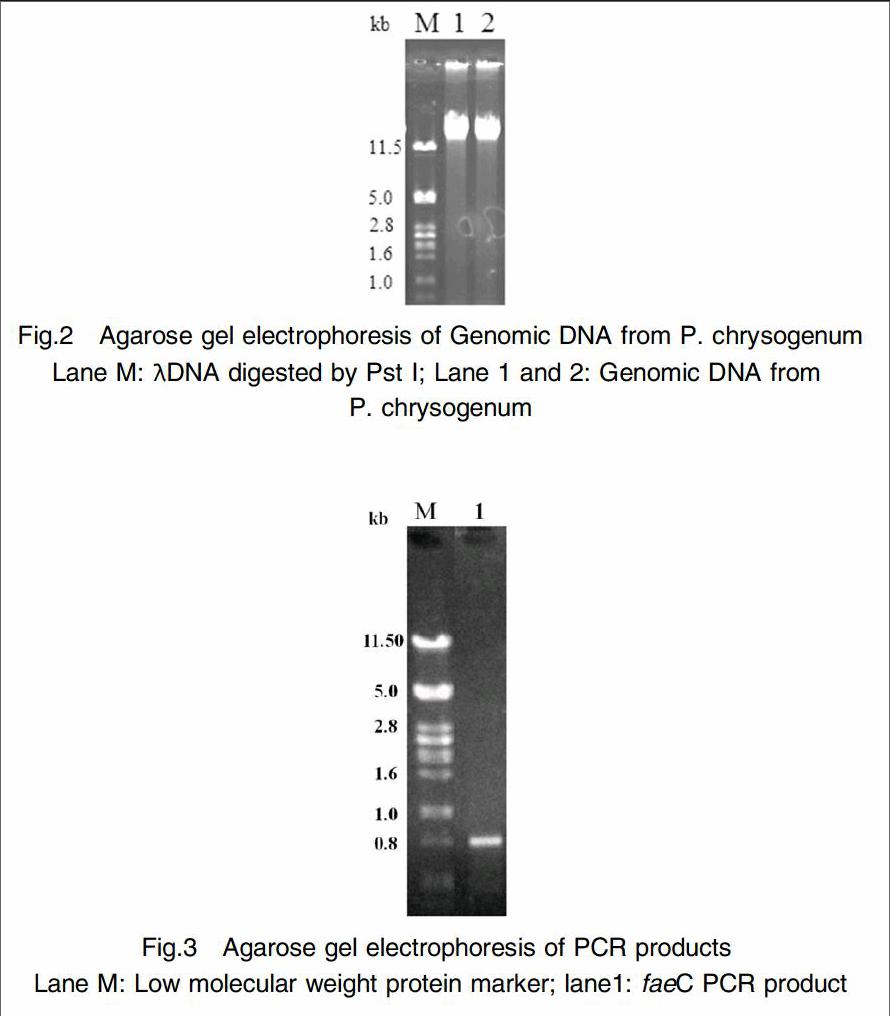
The spores of the P. chrysogenum were inoculated into 100mL PDA liquid medium, then cultured at 30℃, 180 r/min for 1 to 2 days. The thallus was filtrated and the chromosome DNA was extracted by liquid nitrogen grounding method[17]. Based on the feruloyl esterase C gene sequence retrieved by GenBank, two primers: Faec-F1:5-GCGAATTCGCGGCATCTTCGGGCTGC-3 and Faec-R1: 5- GGCCTAGGTCAAGCCGCCATGAAAAACTCC-3 were used to amplify the gene by PCR. Underlined nucleotide indicated the EcoRI and AvrII restriction enzyme sites respectively. The PCR reaction mixture was then subjected to 30 cycles of amplification (30 s at 95℃, 30 s at 60℃, 60s at 72℃), 10min at 72℃ for enlongation. A PCR product without signal peptide was recycled and subcloned into plasmid pMD18-T. The recombiant plasmid introduced into E. coli DH5α and verified by DNA sequencing. The recombiant plasmid was designated as pMD18-PcfaeC.
1.4 Construction of the expression vector
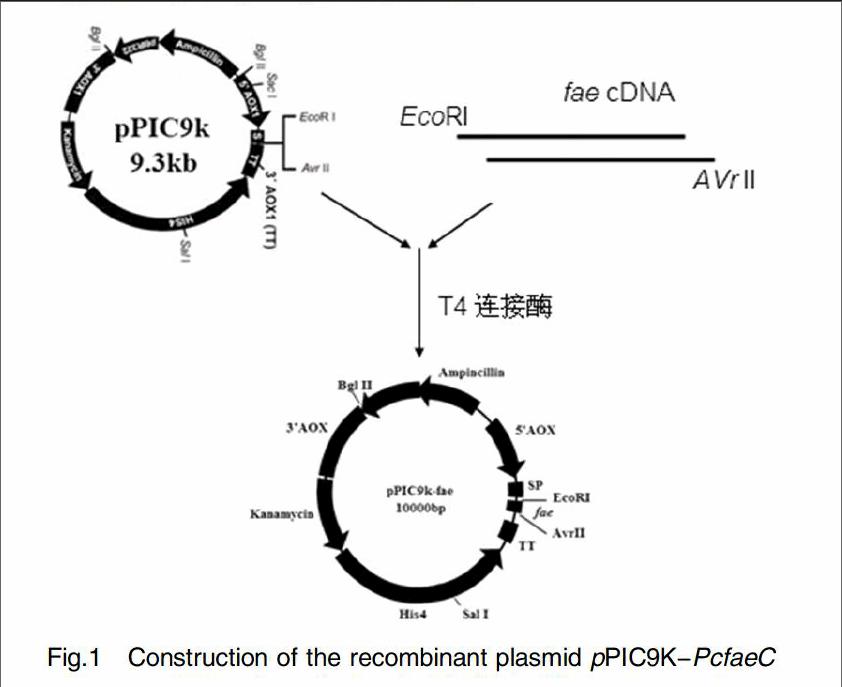
The verified pMD18-PcfaeC plasmid was extracted by Plasmid Maxi Kit, and then digested with EcoRI and AvrII to yield the faeC gene fragment. The purified fragment faeC was then inserted into the pPIC9K plasmid which digested with the same restriction enzyme( Fig. 1). The construction obtained was transformed into E.coli DH5α competent cells and cultivated in LB medium supplemented with 100 ug/mL ampicillin. Positive colonies screened from LB plate was furture identified by PCR and DNA sequencing. The recombiant plasmid was designated as pPIC9K-PcfaeC.
1.5 Screening of the positive colonies and expression
Vectors pPIC9K-PcfaeC and pPIC9K were linerized using BglII restriction enzyme. The recycled and purified fragment was introduced into P. pastoris GS115 by electroporation[18]. The yeast transformants were first screened on MD agar plates after cultured at 30℃ for 2 days. Colonies selected from MD agar plate dibbled on MM agar plates and a new MD plates at 30℃ for 2 days. The corresponding colonies which grow on MD medium while not MM medium was inoculated into 25mL BMGY liquid medium at 30℃, 200 r/min for 2 days and afterwards centrifugated. The resulting pellet was suspended by adding 10mL BMMY medium and furture cultivated at 30℃ on a rotary shaker at 200 r/min for 3 days. Among the 3 days, 50 μl methanol was added to induce the yeast to secret target protein. The induced supernatant was then titrated on the agar medium containing ethyl ferulate and furture culture at 30℃ for 1 hour to verify if transparent zone appeared or not. The positive colonies was screenend based on the appeared transparent zone. The chromosome was extracted according to the P. pastoris brochure provided by invitrogen and furture identified by PCR using the primers: Factor-F: GCTGCTCCAGTCAACACTACAACA; AOX3R: ATGGCATTCTGACATCCTCTTGA; Factor-F/R1; F1/AOX3R and F1/R1 to verify if the faeC gene integrated into the P. pastoris genome, by contrast of the negative control of the empty plasmid pPIC9K.
1.6 SDS-PAGE analysis of the fermentation broth
The crude culture supernatant was obtained by centrifugation of the culture broth at 6000 r/min for 10 min at 4℃. 1 mL of the fermentation broth was taken to the equal volume of the 15% of the trichloroacetic acid. Trichloroacetic acid was used to precipitate the fermentation broth. Afterward, acetone was used to wash the protein precipitation. The protein precipitation was then desiccated and dissolved by PBS. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed to analysis the protein.
1.7 Enzyme activity assay
The standard assay for the feruloyl esterase C enzyme activity was carried out at 40℃ for 10 min in the reaction mixture (500μL) containing 20 mM PBS buffer (pH 6), 0.05mg/mL ethyl ferulate and an appropriate amount of enzyme. The reaction mixture was terminated by adding 500μL of the galcial acetic acid and then entrifugated at 10000r/min for 10min at 4℃. The resulting supernatant was detected by HPLC method. The detailed HPLC conditions were: 4.6×250mm, 5μm GLSciences C8 reversed phase column. Mobil phase: methanol and 1% galcial acetic acid-H2O; flow rate: 1.0mL/min; Column temperature: 35℃; detection wavelength: 322 nm. One unit of the enzyme was defined as the amount of enzyme requied to convert ethyl ferulate to produce 1 μmol ferulic acid at 40℃, pH6.0 per minute.
1.8 Temperature and thermostability of the recombinant enzyme
The optimal temperature was determined by running the standard enzyme assay with appropriate enzyme at different temperatures ranging from 25 to 60℃. The maximal activity was defined as 100% level.
The thermostability was determined by preheating the enzyme in 50 mM sodium phosphate buffer(pH 6) at 30, 40 and 50℃ for different lengths of time, respectively, and the residual activities were measured. The maximal activity was defined as 100% level.
1.9 pH optima and pH stability of the recombinant enzyme
The optimal pH was determined by running the standard enzyme assay with appropriate enzyme in 50 mM various pHs buffer as follows: citrate-phosphate buffer (pH 4-5), sodium phosphate buffer (pH 6-7.5). The maximal activity was defined as 100% level.
The pH stability was determined by pre-incubating the enzyme in various pH conditions at 35℃ for 1 hour, respectively, and the residual activities were measured. The maximal activity was defined as 100% level.
2 Results and Discussion
2.1 Gene cloning and sequence analysis
P. chrysogenum HA4089 chromosome DNA was extracted by liquid nitrogen grounding method (Fig. 2). Based on the chromosome DNA, PCR method was used to amplify the target gene(Fig 3). It is indicated that the PCR product was in good accordance with the size derived from GenBank. The PCR product was validated by gene sequencing, indicating that it comprised one open reading framwork(ORF) and annotated to encode 249 amino acid with a calculated molecular weight of 25.784 kDa and pI 6.14. The protein showed high sequence similarity to its homologous from Penicillium rubens(XP_002563600.1) except that the his24 was substituted for tryosine.
2.2 Expression of the recombinant feruloyl esterase C
As is described in the materials above, the recombinant plasmid pPIC9K-PcfaeC was successfully constructed. The recombinant plasmid pPIC9K-PcfaeC was linerized and introduced into P. Pastoris, 58 transformants were obtained. 7 transformants with relative higher enzyme activity were selected using plate assay when detect the fermentation supernatant. The maximum enzyme activity was found to be 0.36 U/mL using HPLC analysis (Fig. 4)
The transformation efficiency was analyzed by using PCR method based on the transformants genome which introduced into the recombinant plasmid pPIC9K-PcfaeC3 and the empty pPIC9K respectively. The alchol oxidase gene AOX1 on the yeast genome would be substituted by the corresponding fragment of the linerized plasmid pPIC9K-PcfaeC3 and the empty plasmid pPIC9K. The phenotype of the two transformants on the MD plates were all His+Muts. PCR validation results showed that the transformant introduced recombinant plasmid pPIC9K-PcfaeC3 appeared the target band which was in good accordance with the anticipated size by contrast of the negative control. These results indicated that the PcfaeC gene was successfully integrated into the P. pastoris genome.
2.3 SDS-PAGE analysis of the fermentation supernatant
The methanol induced recombinant P. pastoris fermentation broth was sampled per 12 hours to detect the enzyme activity, as described in Materials and Method. The enzyme activity reached its peak after induced 3 days. SDS-PAGE analysis to the No.3 transformant shown that there has a specific band located at 30 kDa by contrast the negative control empty plasmid pPIC9K (Fig. 5). It demonstrated that the target gene was successfully expressed. The actual molecular weight of the recombinant enzyme was relatively larger than theoretical since glycosylation.
Lane M: Low molecular weight protein marker; lane1: Fermentation supernatants from pPIC9K-PcfaeC3/GS115; lane 2: Fermentation supernatants from pPIC9K/GS115 (10μL sample)
2.4 Temperature optima and thermostability
The most optimal temperature was found to 40℃ (Fig. 6). It displayed more than 80% of the maximum activity ranging from 35-45℃. Even at 60℃, The enzyme activity was also retained 44.4% of its optimal activity. Thermostability of the recombinant enzyme was determined by measuring the residual activity after incubation at various temperatures for different lengths of time. The activity of the enzyme was dropped slightly after incubation at 30℃ and 40℃ for 70 min, more than 80% of the activity were retained. While the enzyme activity dropped quickly and lost all its activity when subjected to heat treatment at 50℃ for 20 min (Fig. 7). These results indicated that the enzyme was heat-liable.
2.5 pH optima and pH stability
The optimal pH was found to be 6. It retained beyond 80% of the optimal activity when pH ranges from 5.5-6.5. At pH 4 or 7.5, the activity of the recombinant enzyme was only 30% of the original activity. It can be seen that the enzyme was not suitable for catalyzing at high basid and acid conditions. The pH stability was determined by pre-incubating the enzyme in various pH conditions at 35℃ for 1 hour. The enzyme activity was almost unchanged when incubation the enzyme at pH 5.5-7. We found that the enzyme activity lost most of its activity when subjected to lower and higher pH conditions for 1 hour(Fig. 8). This indicate that the enzyme could not resistant to relatively lower or higher pH conditions.
3 Discussion
The P. pastoris is one of the most successfully foreign protein expression systems. It processes the posttranscriptional modification such as high level foreign protein expression, glycosylation and the formation of the disulfide bond. In this study, the feruloyl esterase C gene was inserted into the pPIC9K vector and successfully expressed in P. pastoris under the regulation of the AOX1 promoter. The feruloyl esterase C was secreted out of the cell under the guidance of the α-factor signal peptide. The α-factor signal peptide was also finally cleaved in the end to generate the mature feruloyl esterase C.
Feruloyl esterase is a key debranching enzyme involved hemicellulase system. It plays an important role in bioresource degration. It is reported that the feruloyl esterase A gene from A. niger[19] had been successfully expressed in P. pastoris, but rarely reported from the source P. chrysogenum. Although we have successfully expressed the feruloyl esterase C from the source P. chrysogenum in P. pastoris, the enzyme activity was relatively low. Site-directed mutagenesis or protein directed evolution technology could be further explored to improve the specific activity and thermostability of feruloyl esterase. Hence, it can lay the foundation for the application of feruloyl esterase in industry.
【References】
[1]Kroon P A, Garcia-Conesa M T, Fillingham I J, et al. Release of ferulic acid dehydrodimers from plant cell walls by feruloyl esterases [J]. Journal of the Science of Food & Agriculture, 1999,79(3):428-434.
[2]Benoit I, Danchin EG, Bleichrodt RJ, et al. Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity [J]. Biotechnology letters, 2008,30(3):387-396.
[3]Wong D. Feruloyl esterase: a key enzyme in biomass degradation[J]. Applied Biochemistry & Biotechnology, 2006,133(2):87-112.
[4]Kroon P A, Faulds C B, Williamson G. Purification and characterization of a novel esterase induced by growth of Aspergillus niger on sugar-beet pulp [J]. Biotechnology & Applied Biochemistry, 1996,23(3):255-262.
[5]Crepin V F, Faulds C B, Connerton I F. A non-modular type B feruloyl esterase from Neurospora crassa exhibits concentration-dependent substrate inhibition [J]. Biochemical Journal, 2003,370(2):417-27.
[6]Vafiadi C, Topakas E, Christakopoulos P. Regioselective esterase-catalyzed feruloylation of L-arabinobiose[J]. Carbohydrate Research, 2006,341(12):1992-1997.
[7]Vafiadi C, Topakas E, Christakopoulos P, et al. The feruloyl esterase system of Talaromyces stipitatus: determining the hydrolytic and synthetic specificity of TsFaeC [J]. Journal of Biotechnology, 2006,125(2):210-221.
[8]Luonteri E, Kroon P A, Tenkanen M, et al. Activity of an Aspergillus terreus α-arabinofuranosidase on phenolic-substituted oligosaccharides[J]. Journal of Biotechnology, 1999, 67(1):41-48.
[9]Bonnina E, Brunel M, Gouy Y, et al. Aspergillus niger I-1472 and Pycnoporus cinnabarinus MUCL39533, selected for the biotransformation of ferulic acid to vanillin, are also able to produce cell wall polysaccharide-degrading enzymes and feruloyl esterases[J]. Enzyme & Microbial Technology, 2001,28(1):70-80.
[10]Crepin V F, Faulds C B, Connerton I F. Functional classification of the microbial feruloyl esterases [J]. Applied Microbiology & Biotechnology, 2004,63(6):647-652.
[11]Garcia B L, Ball A S, Rodriguez J, et al. Induction of ferulic acid esterase and xylanase activities in Streptomyces avermitilis UAH30[J]. Fems Microbiology Letters, 1998,158(1):95-99.
[12]Zhang S, Pei X A, Zhongliu W U. Cloning and Expression of Feruloyl Esterase A from Aspergillus niger and Establishment of Fast Activity Detection Methods [J]. Chinese Journal of Applied & Environmental Biology, 2009, 2009(2):276-279.
[13]Zhang S B, Zhai H C, Wang L, et al. Expression, purification and characterization of a feruloyl esterase A from Aspergillus flavus[J]. Protein Expression & Purification, 2013,92(1):36-40.
[14]Gong Y Y, Yin X, Min-Chen W U, et al. Cloning, Expression and Enzymatic Characterization of Feruloyl Esterase A from Aspergillus usamii [J]. Journal of Food Science & Biotechnology, 2013,32(7):706-712.
[15]Cereghino J L, Cregg J M. Heterologous protein expression in the methylotrophic yeast Pichia Pastoris[J]. Fems Microbiology Reviews, 2000,24(1):45-66.
[16]Sambrook B J, Russell D W. Molecular Cloning: A Laboratory Manual. 3rd edition[M]. Cold Spring Harbor Laboratory Press, 2001.
[17]Al-Samarrai T H, Schmid J. A simple method for extraction of fungal genomic DNA[J]. Letters in Applied Microbiology, 2000,30(1):53-56.
[18]Cregg JM, ed: Pichia Protocols. 2nd edition [M]. Totowa, New Jersey: Humana Press,2007.
[19]Faulds C B, Williamson G. Ferulic acid esterase from Aspergillus niger: purification and partial characterization of two forms from a commercial source of pectinase [J]. Biotechnology & Applied Biochemistry, 1993,17(3):349-359.
[责任编辑:汤静]
猜你喜欢
广州大学学报(自然科学版)(2020年1期)2020-08-03
暨南学报(哲学社会科学版)(2017年5期)2017-11-14
创意与设计(2017年3期)2017-07-31
国际税收(2016年2期)2016-12-29
暨南学报(哲学社会科学版)(2016年12期)2016-11-26
暨南学报(哲学社会科学版)(2016年3期)2016-11-25
暨南学报(哲学社会科学版)(2015年5期)2015-11-14
暨南学报(哲学社会科学版)(2015年2期)2015-11-14
暨南学报(哲学社会科学版)(2015年3期)2015-11-14
暨南学报(哲学社会科学版)(2015年1期)2015-11-14
