
米曲霉酸性蛋白酶Ap的结构分析
2016-05-23 07:30:02李嘉盛曾松荣朱兆静黄小惠韶关学院英东生命科学学院广东韶关512005
食品与生物技术学报 2016年1期
关键词:结构分析
柯 野, 李嘉盛, 曾松荣, 朱兆静, 黄小惠(韶关学院英东生命科学学院,广东韶关512005)
米曲霉酸性蛋白酶Ap的结构分析
柯野,李嘉盛,曾松荣,朱兆静,黄小惠
(韶关学院英东生命科学学院,广东韶关512005)
摘要:米曲霉(Aspergillus oryzae)酸性蛋白酶与部分商品化的蛋白酶相比,具有对大豆蛋白水解效率高且水解产物苦味弱的特性。作者采用RT-PCR技术克隆获得酸性蛋白酶(Ap)基因,结合生物信息学手段,对Ap进行分析与预测。结果表明:Ap基因对密码子使用频率具有偏好性,特别是第3位密码子对GC碱基的使用频率高,达到74%;编码404个氨基酸残基。Ap属于胞外天冬氨酸蛋白酶。以海枣曲霉(A. phoenicis)的酸性蛋白酶(PDB code:1IBQ)作为模板同源建模结果可知,Ap分子至少能与2个Zn2+结合,内含1个二硫键、163个氢键、35个盐碱。该酶与1IBQ相比空间结构总体相似,部分重要位点(如flap环、转角和ψ-loops)的氨基酸残基较为保守。
关键词:米曲霉;酸性蛋白酶;结构分析
蛋白酶是一类重要的工业酶,占整个酶制剂总量的60%以上,广泛应用于医药、皮革、食品和化工等多种行业[1]。目前商品化的蛋白酶制剂主要来源于细菌和植物;而霉菌源蛋白酶主要来自于毛霉和曲霉,且商品化相对较少。霉菌蛋白酶对植物蛋白的水解效率高、无苦味,水解产物具有多种生理活性,具有其他商品化蛋白酶无可比拟的优势而日趋受到关注[2]。
米曲霉(Aspergillus oryzae)是生物技术中重要的一种曲霉。该菌广泛应用于传统的酒类、酱油和豆酱等食品发酵工业;主要是由于米曲霉能产生多种蛋白酶,这些蛋白酶对大豆蛋白的水解效率高、水解产物苦味少以及对独特风味起关键作用[1,3],对提高食品发酵行业原料蛋白质的氨基酸态氮转化率和利用率起重要作用;因此,米曲霉蛋白酶受到科研工作者的普遍关注。目前,国内外学者的研究主要集中于利用传统的育种手段和优化发酵条件方式来提高米曲霉的产蛋白酶量;或分离纯化蛋白酶,对其酶学性质方面的研究[4]。对于蛋白酶的分子生物学方面的研究,主要体现在对米曲霉全基因组的测序;据2007年全基因组的测序结果预测可知,该菌一共编码134个蛋白酶,其中65个内肽酶、69个外肽酶,只有少数几个蛋白酶的功能与性质有所掌握,而绝大多数蛋白酶的结构与功能方面的研究鲜见报道。因此,作者根据Genbank中报道的米曲霉酸性蛋白酶(Ap)的基因序列,对该基因进行RTPCR克隆及测序;利用生物信息学对该基因及其推导编码的酶学性质、结构与功能等方面进行分析,以期对该酶的结构功能、外源表达和定向改造等方面的研究提供参考。
1 材料与方法
1.1试验材料
1.1.1菌株米曲霉(A. oryzae):购自上海迪发酿造生物制品有限公司。
1.1.2工具酶和试剂PCR扩增引物:购自生工生物工程(上海)股份有限公司;RT-PCR试剂:购自宝生物(大连)有限公司;Trizol试剂:购自Invitrogen公司;其他化学试剂均为国产分析纯。
1.2试验方法
1.2.1米曲霉的培养在麸皮固体培养基平板上先铺上灭菌后的玻璃纸,然后接种米曲霉,25.0℃培养48 h后收集菌丝体。麸皮培养基成分的具体组成如下(质量分数):0.74% KH2PO4,0.06% FeSO4· 7H2O,1.56%蛋白胨,0.23%麦芽糖,96.41%麸皮,培养基初始含水量为1.0~1.5 mL/g。
1.2.2 RNA的提取将收集的菌丝体浸泡在液氮中进行研磨至粉末,根据Trizol试剂盒的操作手册提取并鉴定总RNA质量,将提取的总RNA放置于-70℃冰箱中保存备用。
1.2.3 cDNA的合成利用宝生物(大连)有限公司的反转录试剂盒,按照试剂盒的操作手册,对RNA反转录生成cDNA。
1.2.4 Ap基因的克隆由Genbank数据库中报道的米曲霉Ap基因序列(登录号:XM_001824123),利用Oligo 6.0软件设计引物,上游引物:5’-CTATGGTTATCTTGAGCAAAG-3’,下游引物:5’-ATTAGTCAGGCATTTAAG-3’,以1.2.3的cDNA作为模板,采用上下游引物进行PCR反应。反应程序如下:94℃预变性4 min;94℃变性15 s,50℃退火15 s,72℃延伸2.5 min,30个循环;然后72℃保温5 min。电泳鉴定PCR产物,对PCR产物回收加A后,克隆至载体pMD18-T上,化转于大肠杆菌DH5α菌株中,筛选阳性转化子测序确认。
1.2.5 Ap基因的分析利用Bio-Edit 7.0软件进行序列比对,http://www.expasy.org/中的部分程序对酶序列的功能位点进行初步分析,http://www.cbs. dtu.dk/services /TargetP/的信号肽分析,http:// swissmodel.expasy.org/进行空间结构的预测,http:// swift.cmbi.ru.nl/ servers/html/分析拉曼图对结构合理性的评价,Swiss -Pdb Viewer 4.0.3和Discovery studio 2.5等软件进行酶空间的比对分析,http:// swift.cmbi.ru.nl/servers/html/的程序对酶的氢键数、盐键数和溶剂可及表面积等方面的预测分析。
2 结果与分析
2.1米曲霉的总RNA提取及Ap基因全长cDNA的克隆结果
米曲霉的总RNA提取结果见图1。图1可见,28s rRNA和18s rRNA条带清晰可见,28s rRNA的宽度和亮度分别约为18s rRNA亮度的1倍和2倍,且RNA未见明显降解;该结果表明提取的RNA质量较高,达到RT-PCR的质量要求。
利用Ap基因的引物和以cDNA为模板,获得预期的PCR产物(见图2)。该PCR产物测序结果表明,Ap核苷酸序列与GenBank数据库报道的同源性达到99 %以上,仅核苷酸T981突变为C;而相应的氨基酸残基未突变。利用http://www.kazusa.or.jp/ codon/对A. oryzae的12 388条CDS序列的密码子使用频率分析可知,该菌对密码子使用存在偏好性,即第1位、第2位和第3位的密码子GC使用频率分别为56.53 %、43.87 %和55.88 %;对Ap基因而言,第1位、第2位和第3位的密码子GC使用频率分别为58.0%、46.0%和74.0%。在一般情况下,密码子在同一生物存在相似的密码子偏好性;该结果也表明Ap基因的密码子使用偏好性与米曲霉中基因密码子使用偏好性较一致。
2.2 Ap的一级结构分析
Ap是由404个氨基酸残基组成,其中Met和Cys残基数最少(2个),Gly残基数最多(44个)。G、S、T、C、Y、N和Q极性氨基酸为162个,A、V、L、I、F、W、M和P非极性氨基酸残基数为167个;该结果表明极性和非极性氨基酸残基的含量较一致。氨基酸残基序列中1-20氨基酸序列为信号肽,21-78氨基酸序列为前导肽,79-404氨基酸序列为成熟肽;成熟肽的理论等电点为4.03,该酶属于胞外的天冬氨酸蛋白酶。利用http://www.expasy.org/prosite对活性位点的预测可知,Ap的活性位点残基为D111和D293。

图1 米曲霉的总RNA琼脂糖凝胶电泳Fig. 1 Agarose gel electrophoresis of isolated RNA from A. oryzae

图2 Ap基因的PCR产物电泳Fig. 2 Electrophoresis of PCR products of Ap gene
2.3 Ap的同源建模及二级结构分析
用Ap的成熟肽序列(即Ap全长氨基酸序列的第79-404肽链)与海枣曲霉(A. phoenicis)酸性蛋白酶的序列(该序列是PDB数据库(http://www.rcsb. org/pdb/home/home.do)中提供的成熟蛋白酶晶体结构的全长氨基酸序列,该酶晶体结构的编号为1IBQ)同源性达到73.375%,(见图3);据序列同源性达到30%以上可以进行同源建模的原则,这表明1IBQ可以作为Ap的模板进行同源建模。模拟的Ap结构见图4,利用Ramachandran对该模型分析,结果表明其主链ψ和φ角均在合理范围之内,这表明构建的3-D结构合理。
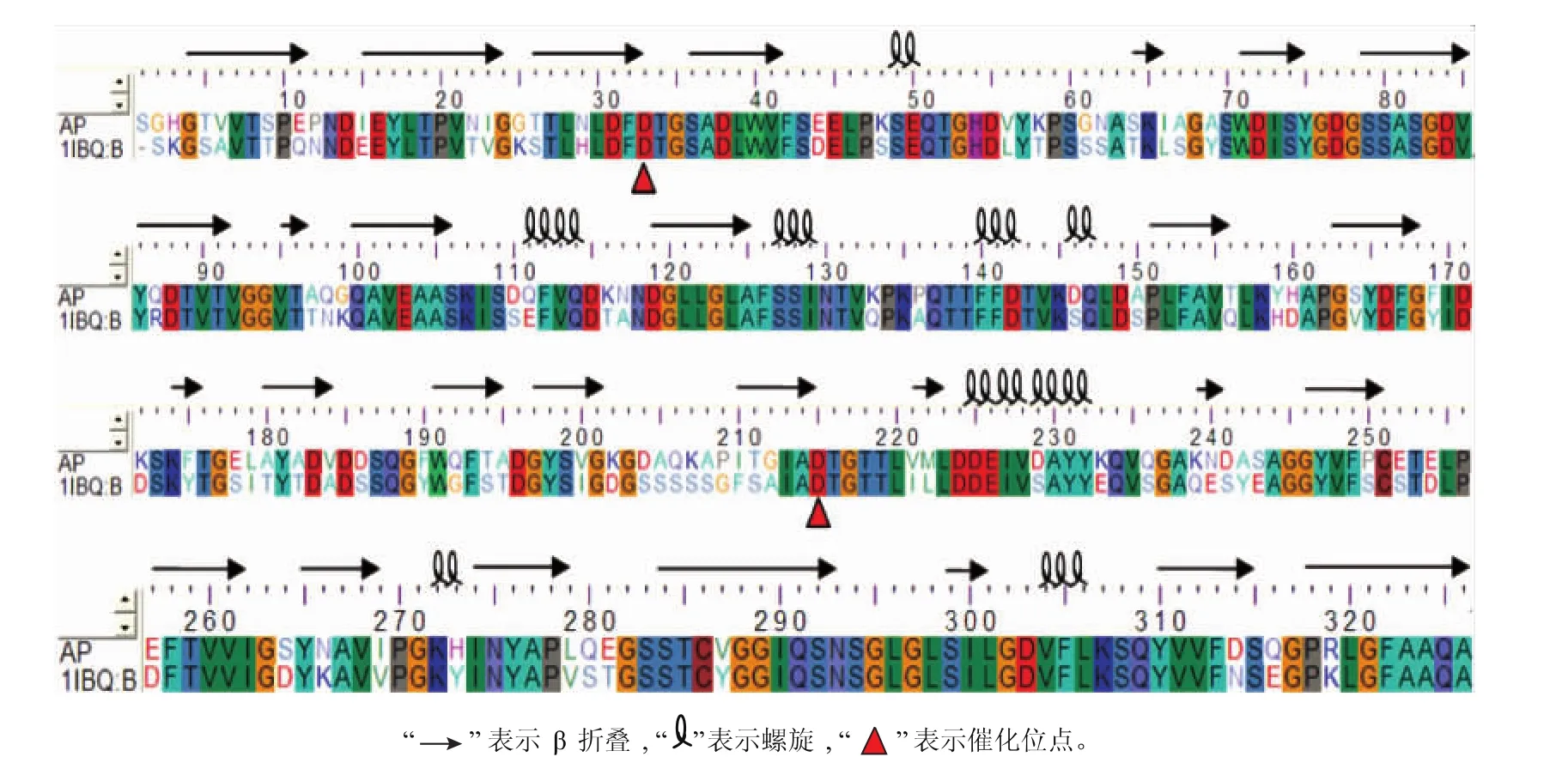
图3 Ap与海枣曲霉酸性蛋白酶(PDB code:1IBQ:B)的序列比对及二级结构分析Fig. 3 Comparison of sequences and secondary structures of Ap and acid protease(PDB code:1IBQ.B)from A. phoenicis
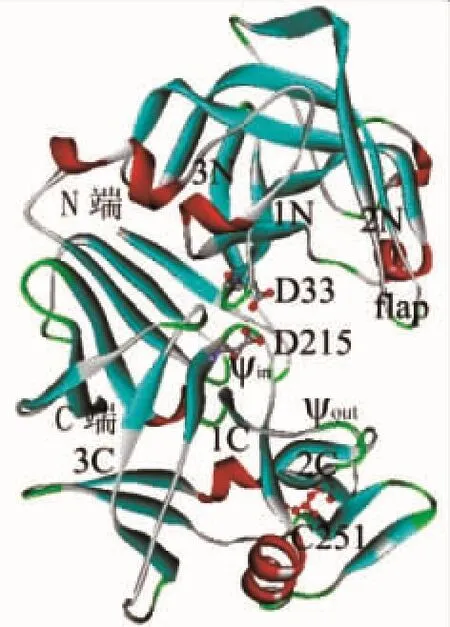
图4 Ap的3-D结构模型Fig. 4 Overall 3-D structure model of Ap
由图4可见,Ap是由两个明显的C-端和N-端结构域的β-barrel构成。N-端结构域和C-端结构域中均分别包含3个β折叠,即1N、2N、3N、1C、2C 和3C;N-端结构域与C-端结构域是由6个β折叠链连接在一起,并且两结构域之间形成的cleft是底物结合区域[5],此外Ap中还包含3个310-螺旋和5 个α螺旋。活性位点残基D33(根据Ap的成熟肽序列编号,下同)和D215分别位于N-端结构域与C-端结构域上。由Y75-G78残基片段形成的β发夹结构为flap结构,该结构保护活性残基免受溶剂的攻击[6]。Ap与模板1IBQ的结构比较可知,虽然这两种蛋白酶的结构大体相似,但是部分二级结构的长短和连接二级结构间的loop环等方面的残基具有差异,这些差异可能是导致两种酶性质不同的原因之一[7]。
2.4 Ap保守残基的分析
通过对多种蛋白酶(如PDB code:1IBQ、3APP、4APE和2APR等)的分析可知:在这些酶中,部分位点的氨基酸非常保守,且这些位点主要位于N-端结构域,少数位于C-端结构域;如24、35、76、78、83、120、123、168、177、217和302位点均是G,该G对酶二级结构中的转角构象起重要作用。33-35片段和215-217片段形成保守的活性位点“DTG”Motif结构;在酶的空间结构中,T34和T216残基的侧链羟基分别与对面活性位点“DTG”Motif结构中活性残基D的上一个残基主链上的N或O形成氢键,因此,该氢键链接两个ψ-loops环,维持和稳定活性位点区域的局部结构具有重要作用,该结构被称为“fireman’s grip”结构[6]。Y17、W40、S60、Y75、D88、Q100和A323等位点较为保守,这些残基间通过氢键作用连接或调控相邻的loop环或链,具有维持局部结构稳定的作用,如N-端结构域β链上的Y17侧链上的H与邻近β链上K158主链上的羰基O形成了氢键,这个氢键维持两个β链间的稳定性起到重要作用。
2.5 Ap中Zn2+和二硫键分析
Zn2+离子在较多的酶中具有催化或维持结构稳定的作用[8]。对于1IBQ而言,该酶结合了4个Zn2+结合(见图5);其中2个Zn2+(即Zn1和Zn2)位于酶分子的内部,另外2个Zn2+(即Zn3和Zn4)位于酶分子表面,该两个Zn2+调控对称的两个酶分子间的接触。对于Zn1而言,Zn2+位于1IBQ结构中的H28、D55和D119残基之间,并且Zn2+与D55侧链上的O结合;在Ap中,D55和D119的残基均保守,N28残基的侧链基团比1IBQ中的H残基的侧链基团小,占用的空间位置也就相应的减少,因此在该空间中能容纳Zn2+,Ap在Zn1位点能够结合Zn2+(见图5 b)。对于Zn2而言,Zn2+位于模板1IBQ的活性残基D33和D215间,Zn2+与D214侧链上的O结合;在Ap中,活性位点D32和D215残基为保守残基,空间大小和距离均与1IBQ一致,这表明Ap在Zn2位点能结合Zn2+。对于Zn3而言,Zn2+位于1IBQ 的D149与Ser2之间,且Zn2+与D149的残基侧链相结合;对于Ap,在位点1具有S残基,但空间结构的预测中未含S1残基,因此无法判断是否结合Zn2+。对于Zn4而言,Zn2+位于1IBQ的E233位点;在Ap中为K残基,K残基的侧链基团比E长,占据了Zn2+空间,因此Ap可能Zn4位点无Zn2+结合。
在一般情况下,二硫键利于酶的热稳定性与结构稳定性[9]。在1IBQ中C251和C281残基间形成二硫键;在Ap中该2位点也有C残基,且其与1IBQ的空间结构一致,因此Ap中该2个C之间形成一个二硫键。
2.6 Ap氢键和盐键的分析
酶中的氢键对酶整体结构的稳定性具有极其重要的作用;相对而言,盐键更有助于局部结构的稳定性[10]。利用http://swift.cmbi.ru.nl/servers/html/中的Optimal Hydrogen Bonding Network算法可知,1IBQ B的氢键数为164个,Ap为163个。1个盐键的定义:以相距为7.0Å以内且带相反电荷的2个残基之间可形成1个盐键,H残基定义为带正电荷;因此,1IBQ B的盐键数为41个,Ap为35个;在这些盐键中,大部分位点的盐键较保守,对酶的稳定性起重要的作用,如位于N端结构域的D14残基和位于C端结构域的K307残基间形成1个盐键,该盐键对两结构域间的相对稳定性起到一定的作用。由上可知,两种酶间的氢键和盐键的位置与数量具有一定的差异,这种差异可能是导致两种酶稳定性不同的原因之一。
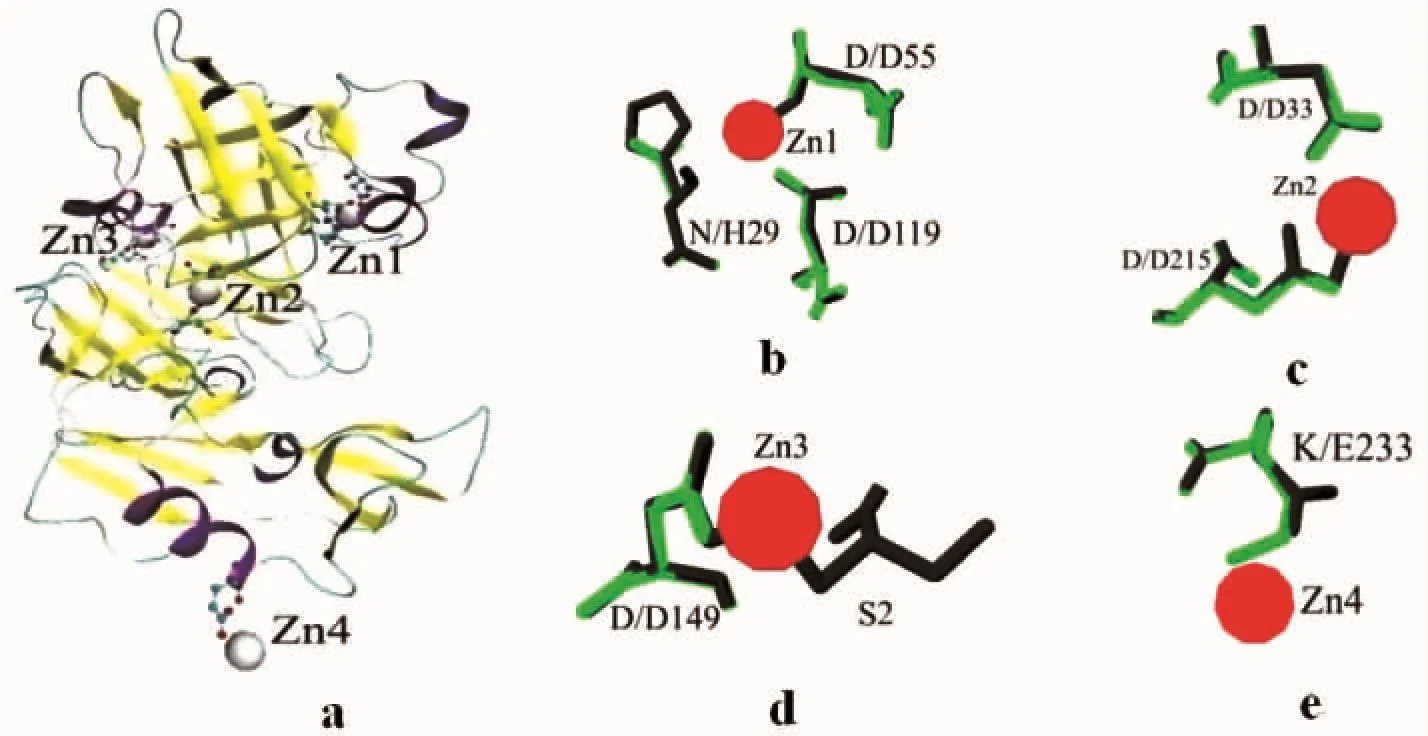
图5 Ap与海枣曲霉酸性蛋白酶的Zn2+结合位点分析Fig. 5 Analysis of the zinc binding sites of Ap and acid protease(PDB code:1IBQ.B)from A. phoenicis
2.7 Ap活性位点的溶剂可及表面积的分析
天冬氨酸蛋白酶的催化机制是:利用一个活性催化残基D侧链的O原子和溶液中的H2O结合后激活H2O分子,激活后的H2O对底物肽链骨架中羰基的C原子进行亲核攻击,另一个催化残基D对底物的质子化导致底物肽链的切断[11]。溶剂可及表面积表明酶与底物和H2O等的接触情况,对酶的催化效率和催化环境的适应性等方面具有重要的影响[12]。对两种酶的活性位点残基的溶剂可及表面积计算可知:两种酶活性位点残基主链的溶剂可及表面积均为0;而侧链的溶剂可及表面积的差异较大,Ap的活性位点残基的侧链溶剂可及表面积均高于1IBQ,这就表明Ap的侧链基团更易与H2O结合,可能更利于亲核攻击,提高酶的催化活性。
2.8 Ap底物结合区域的ψ-loop结构分析
酶的底物结合区域与底物结合后,才能对底物进行水解切割;因此,底物结合区域对酶的肽键选择性、水解特性和水解效率等方面起关键性的作用[13]。由图3-4可知,两个灵活的ψ-loops分别均是Ap和1IBQ B的底物结合区域S1’-S2的重要组成部分;这两个ψ-loops均位于酶的1C区域,每个ψloop均包含2个β链。一个ψ-loop称为ψin环,主要由211-223位点的残基片段构成;活性位点“DTG”Motif结构位于ψin结构的转角。在两种酶的比较可知,转角部分的残基保守,而β链顶部的2个残基具有突变,即1IBQ是S211-A212和I221-L222,在Ap中分别突变为TG和VM,而该几个突变残基的空间结构相似,且离活性位点相对较远,对酶的性质影响较小。另一个称为ψout环,主要由完全保守的289~302位点的残基片段构成,该环突出于酶表面,部分覆盖了酶的活性cleft,环中的L296 和L298残基对S1’-S2底物结合区域的构型起重要的作用。
3 结语
克隆获得米曲霉的酸性蛋白酶(Ap)基因,然后对Ap的结构进行了预测与分析。结果表明:Ap基因对密码子使用频率具有明显的偏好性。Ap属于胞外天冬氨酸蛋白酶,且Ap分子至少能与2个Zn2+结合,含1个二硫键、163个氢键、35个盐碱。该酶的部分重要位点(如flap环、转角和ψ-loops)的氨基
参考文献:酸残基较为保守,研究可为该酶的结构功能、异源表达和定向改造等方面的研究提供参考。
[1]A Sumantha,C Larroche,A Pandey. Microbiology and industrial biotechnology of food-grade proteases:a perspective[J]. Food Technology and Biotechnology,2006,44(2):211-220.
[2]潘进权.蛋白酶脱除大豆蛋白水解物苦味的研究进展[J].食品研究与开发,2011,32(5):167-171. PAN Jinquan. Research progress on the debittering of soy protein hydrolysate with proteases[J]. Food Research And Development,2011,32(5):167-171.(in Chinese)
[3]T Nakadai,S Nasuno,N Iguchi. The action of peptidases from A.oryzae in digestion of soybean proteins[J]. Agricultural and Biological Chemistry,1972,36(2):261-268.
[4]J Chutmanop,S Chuichulcherm,Y Chisti,et al. Protease production by Aspergillus oryzae in solidstate fermentation using agroindustrial substrates[J]. Journal of Chemical Technology and Biotechnology,2008,83(7):1012-1018.
[5]SW Cho,NJ Kim,MU Choi,et al. Structure of aspergillopepsin I from Aspergillus phoenicis:variations of the S1'-S2 subsite in aspartic proteinases[J]. Acta Crystallographica Section D(Biological Crystallography),2001(57):948-956.
[6]JB Cooper,G Khan,G Taylor,et al. X-ray analyses of aspartic proteinases. II. Three-dimensional structure of the hexagonal crystal form of porcine pepsin at 2.3 Å resolution[J]. Journal of Molecular Biology,1990,214(1):199-222.
[7]JS Roland,M de V William,A M L Jack,et al. Homology modelling and protein engineering strategy of subtilases,the family of subtilisin-like serineproteinases[J]. Protein Engineering Design&Selection,1991,4(7):719-737.
[8]IL Alberts,K Nadassy,SJ Wodak. Analysis of zinc binding sites in protein crystal structures[J]. Protein Science,1998,7(8):1700-1716.
[9]Han Zhen-lin,Han Shuang-yan,Zheng Sui-ping,et al. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface[J]. Applied Microbiology and Biotechnology,2009,85(1):117-126.
[10]J Arnórsdottir,M M Kristjánsson,R Ficner. Crystal structure of a subtilisin-like serine proteinase from a psychrotrophic vibrio species reveals structural aspects of cold adaptation[J]. Federation of European Biochemical Societies Journal,2005,272(3):832-845.
[11]S Chitpinityol,MJC Crabbe. Chymosin and aspartic proteinases[J]. Food Chemistry,1998,61(4):395-418.
[12]AH Deng,J Wu,GQ Zhang,et al. Molecular and structural characterization of a surfactant-stable high-alkaline protease AprB with a novel structural feature unique to subtilisinfamily[J]. Biochimie,2011,93(4):783-791.
[13]LM Liang,ZH Meng,FP Ye,et al. The crystal structures of two cuticle-degrading proteases from nematophagous fungi and their contribution to infection against nematodes[J]. Official Publication of the Federation of American Societies for Experimental Biology,2010,24(5):1391-1400.
Structural Analysis of Acid Protease ( Ap) from Aspergillus oryzae
KE Ye,LI Jiasheng,ZENG Songrong,ZHU Zhaojing,HUANG Xiaohui
(Yingdong college of life sciences,Shaoguan University,Shaoguan 512005,China)
Abstract:Compared with some commercial proteases,acid protease(Ap)from Aspergillus oryzae has high hydrolysis efficiency and produces soy hydrosates with weaker bitterness. In order to uncover the structural characteristics,Ap gene from A. oryzae was cloned by RT-PCR technology,and then was analyzed with bioinformatics technology. Results showed that Ap gene had a preference for codon usage,especially for the third codon with the 74% usage frequency of GC base,and encoded 404 amino acid residues. Ap was an extracellular aspartic proteinase. According to the structure obtained by homology modeling with acid protease from A. phoenicis(PDB code:1IBQ),Ap was found to possess at least two Zn2+binding sites,one disulfide bond,163 hydrogen bonds,35 salt bonds,and two solvent accessible surfaces of D33(4.7849Å2)and D215(3.5941Å2). Though the spatial structures of Ap and 1IBQ were similar,residues of some important sites(such as flap ring,angle and ψ-loops)were conservative.
Keywords:Aspergillus oryzae,acid protease,structural analysis
作者简介:柯野(1977—),男,四川泸县人,理学博士,副教授,主要从事微生物学与酶工程的研究。E-mail:keye518@163.com
基金项目:广东省科技计划项目(2011B010500018);韶关市科技专项资金资助项目(2013CX/K83);韶关学院大学生创新创业训练计划省级立项项目(1057613-003);韶关学院大学生科技创新培育项目(2015-6)。
收稿日期:2014-08-21
中图分类号:Q 816;Q 71
文献标志码:A
文章编号:1673—1689(2016)01—0095—06
猜你喜欢
中国科技博览(2016年23期)2016-12-09 09:44:41
智富时代(2016年12期)2016-12-01 14:57:24
文艺生活·中旬刊(2016年10期)2016-11-04 04:44:49
中国科技博览(2016年17期)2016-08-26 23:38:54
戏剧之家(2016年10期)2016-06-18 12:31:11
科技视界(2016年13期)2016-06-13 08:03:44
现代经济信息(2016年6期)2016-05-31 21:17:41
中国科技博览(2016年7期)2016-04-25 20:43:07
中国科技博览(2016年9期)2016-04-25 05:40:11
商(2016年9期)2016-04-15 08:11:21
