
In flammasomes and Atherosclerosis
2016-05-23 01:59:14VallurupalliMDYaoDaiMDPhDandMehtaMDPhD
S. Vallurupalli, MD, Yao Dai, MD, PhD and J. L. Mehta, MD, PhD
1Division of Cardiology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA
2Central Arkansas Veterans Healthcare System, Little Rock, AR, USA
Introduction
Atherosclerosis is an in flammatory disorder.In flammation plays a major role in the initiation and progression of atherosclerosis and leads to potentially life-threatening clinical manifestations via plaque rupture [1]. In flammasomes represent a distinct form of innate immunity. These cytosolic protein complexes are formed in response to offending proteins and lead to the production of proin flammatory enzymes such as caspase 1, resulting in activation of proin flammatory cytokines such as IL-1β and IL-18; their activation results in cellular apoptosis[2]. Since the discovery of in flammasomes in 2002,there has been an explosion in our knowledge of the importance of in flammasomes in chronic in flammatory diseases, especially atherosclerosis.
The goal of the current review is to present the current evidence for the role of in flammasomes,especially NLRP3, in atherosclerosis.
Overview of In flammation in Atherosclerosis
In flammation plays an important role in all phases of atherosclerosis – from initiation to its clinical manifestations such as plaque rupture [1, 3, 4]. When the normal endothelium is exposed to an atherogenic milieu, there is production of reactive oxygen species and reduced availability of nitric oxide. This results in expression of cell adhesion molecules,such as vascular cell adhesion molecule 1 and selectins, which recruit blood monocytes and lymphocytes into the atherosclerotic plaque. Selectins and integrins mediate increased adhesion of leukocytes to the activated endothelial cells. Under the in fluence of chemoattractant proteins such as monocyte chemoattractant protein 1 and macrophage colony stimulating factor, the adherent monocytes migrate into the intima and transform into macrophages.These macrophages transform into foam cells by uptake of modi fied LDL particles such as oxidized LDL (ox-LDL). T lymphocytes that are recruited into the plaque secrete cytokines and growth factors that promote the migration and proliferation of smooth muscle cells (SMCs). Repeated cycles of apoptosis and cell death lead to accumulation of cellular debris with enlargement of the lipid core,and the atheromatous plaque grows. Medial SMCs and foam cells secrete matrix metalloproteinases,which degrade elastin and collagen. This leads to weakening of the plaque, especially at its shoulders,which makes it prone to rupture. When the plaque ruptures, the presence of tissue factor promotes local thrombus formation and leads to the clinical presentation of acute coronary syndromes. This simpli fied explanation of the atherosclerotic process underscores the vital role played by in flammation. Current thinking suggests that atherosclerosis is a result of such “sterile” in flammation.
Overview of In flammasomes
In flammation is mediated by two major pathways:innate and adaptive immunity. Innate immunity provides a “fast and blunt” response to proin flammatory stimuli, while the response is “slower and more precise” with adaptive immunity. In flammasomes are multimeric cytosolic proteins that assemble when molecular patterns that are perceived as a threat are recognized by germline-encoded pattern recognition receptors (PRRs) [5]. These molecular patterns could be either associated with pathogens (pathogen-associated molecular patterns) or proteins associated with cell damage (damage- associated molecular patterns) [6]. PRRs are expressed both in the cell membrane (e.g., Toll-like receptors and C-type lectin receptors) and in the cytoplasm (e.g.,Nod-like receptors) [7].
Figure 1 describes the basic structure of the four well-described in flammasomes (NLRP1, NLRP3,NLRC4, and AIM2). The multimer consists of three basic structural units: (1) a PRR (pathogenassociated molecular pattern or damage-associated molecular pattern), (2) an effector (IL-1β-and IL-18-processing system, i.e., caspase 1), and (3)a coupling protein (the adaptor protein apoptosisassociated speck-like protein containing a carboxy-terminal caspase activation and recruitment domain [CARD], ASC) [9]. ASC is common to all in flammasomes (except NLRP4, which recruits procaspase 1 using CARD alone) and contains two death-fold domains – a pyrin domain and a CARD.The pyrin domain interacts with PRRs and triggers the formation of large ASC multimers. CARD (present both in ASC and in NLRC4 and NLRP1, where it directly recruits procaspases 1 and 5) initiates the recruitment of procaspase 1 to the in flammasome and mediates its cleavage to caspase 1. This oligomerization requires ATP and low intracellular potassium concentrations [10]. The end product,caspase 1, is a cysteine protease that mediates a form of cell death called “pyroptosis” and proteolytically cleaves pro-IL forms to active IL-1β and IL-18 [5]. A comprehensive description of these in flammasomes is available elsewhere. Of these,NLRP3 will form the basis of this review because of its role in atherosclerosis.
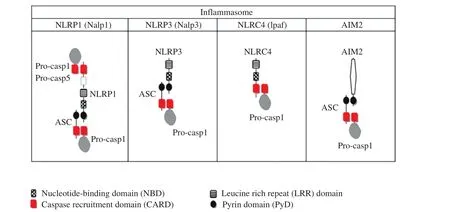
Figure 1 Basic Structure of Four Well-Characterized In flammasomes.Reproduced from [8] with permission from Dr. Hiroko Tsutsui.
NLRP3 In flammasome and Atherosclerosis
The NLRP3 in flammasome is the most extensively studied in flammasome. It has been implicated in the pathogenesis of several disorders, such as gout,rheumatoid arthritis, type 2 diabetes mellitus, and atherosclerosis [5]. Activation of NLRP3 is a twostep process as illustrated in Figure 2. A priming step consists of activation of nuclear factor κB by activation of cell membrane PRRs such as Toll-like receptor 4 [8]. This leads to induction of proforms of IL-1 family cytokines (pro-IL-1β and pro-IL18)as well as NLRP3, and sets the stage for the second step. The second step consists of the phagocytosis of sterile crystalline substances by lysosomes,which transform into phagolysosomes and result in production of cathepsin B; this process initiates the oligomerization of the NLRP3 in flammasome and caspase 1 production [11, 12]. Caspase 1 cleaves the pro-IL-1 forms to active IL-1β and IL-18. IL-1β is often considered the “gatekeeper of in flammation” [13]. Through activation of IL-1 type I receptor, it mediates a proin flammatory state characterized by increased production of inducible nitric oxide synthase, endothelin 1, and other proin flammatory chemokines, cytokines, and adhesion molecules. This results in macrophage activation as well as endothelial and SMC proliferation, leading to progression of atherosclerosis. The role of IL-1β in atherosclerosis has long been established [14,15]. In apolipoprotein E (ApoE)-de ficient mice,lack of IL-1β is the basis for reduction in the severity of atherosclerosis [15]. Carotid ligation induces less neointimal thickening in IL-1β-de ficient mice than in controls [14]. IL-18 is widely considered a circulating biomarker of atherosclerosis and enhances atheromatous plaque progression through interferon-γ production [16]. However, it took the discovery of in flammasomes to understand how certain cardiac risk factors such as hyperlipidemia result in increased IL production.
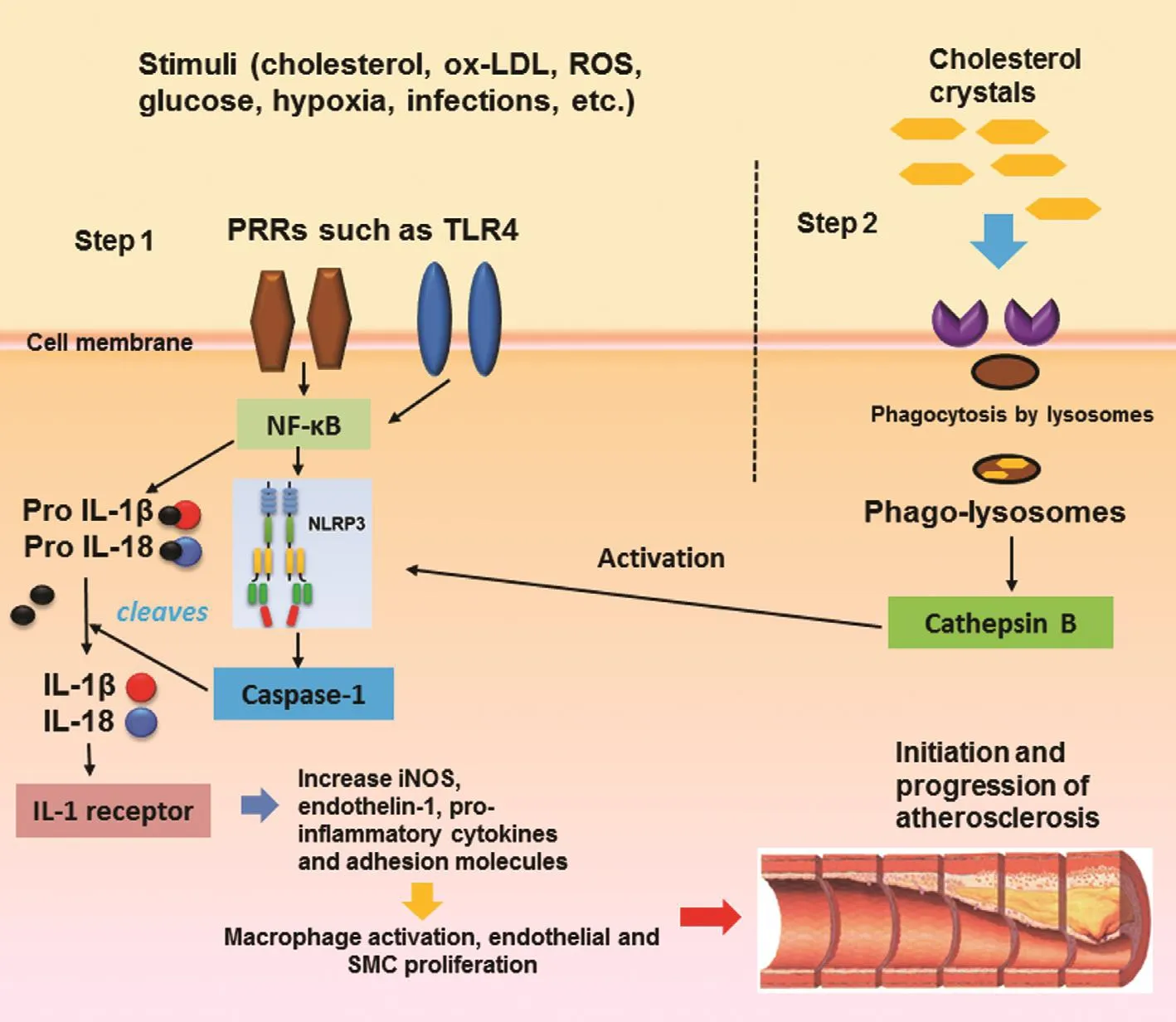
Figure 2 NLRP3 Activation in Atherosclerosis is a Two-Step Process.The priming step (step 1) leads to production of nuclear factor κB and proforms of IL-1 and IL-18. The second step is mediated by phagocytosis of substances such as crystalline cholesterol and leads to production of caspase 1, which cleaves the inactive forms to active forms.
Crystalline substances such as urate crystals, silica, and alumina have long been known to initiate the second step of NLRP3 in flammasome activation. Duewell et al. [11] made the seminal observation that cholesterol crystals lead to activation of in flammasomes. Recent studies from our laboratory suggest that xanthine oxidase, the action of which results in the formation of uric acid, can induce foam cell formation in macrophages and SMCs,and in this process activation of lectin-type ox-LDL receptor 1 (LOX-1), a and C-type lectin receptor,plays a crucial role (unpublished observations).
Both extracellular and intracellular cholesterol crystals can result in NLRP3 activation. Exposure of human macrophages to ox-LDL has long been known to be a potent activator of NLRP3 [17]. This is mediated by endocytosis of ox-LDL by the scavenger receptor CD36 [18]. Ox-LDL then directly primes NLRP3 activation by production of pro-IL forms (priming signal) as well as nucleates to intracellular cholesterol crystals, resulting in activation of NLRP3 (second signal). The process of formation of intracellular cholesterol crystals in human macrophages is now well understood [19]. Cholesterol exists mostly in the arterial wall (as LDL particles) in a esteri fied form. When LDL is oxidized and assimilated by macrophages, cholesterol ester hydrolase converts esteri fied cholesterol to free cholesterol, whereas acyl coenzyme A cholesterol acyltransferase 1 converts free cholesterol back to esteri fied cholesterol, thus maintaining an equilibrium. Disruption in cholesterol homeostasis leads to the formation of crystals of free cholesterol within the macrophage and activates in flammasomes.
Recent studies from our laboratory suggest that NLRP3 mediates the deleterious effects of the activation of LOX-1 [20, 21]. In cultured human macrophages, LOX-1 inhibition with a binding antibody or small interfering RNA resulted in decreased expression of the NLRP3 in flammasome and thus inhibited reactive oxygen species generation,autophagy, and mitochondrial DNA damage [20].This is con firmed by reduced mitochondrial DNA and reduced NLRP3 activation in LOX-1-knockout mice (Figure 3) [21].
In addition to cholesterol crystals, other lipid and nonlipid signals modulate NLRP3-mediated in flammation, especially in the context of atherosclerosis:
1. Electronegative LDL, a modi fied LDL fraction,has been shown to promote the release of IL-1β through both the priming step and the activation step of NLRP3 [22].
2. ATP is secreted by activated or injured endothelial cells, leukocytes, and platelets, and then acts in a paracrine manner to transduce sterile in flammatory signals [23]. This transduction is likely mediated by NLRP3. When exposed to ATP,macrophages from wild-type mice demonstrate NLRP3 activation along with increased lipid deposition in lysosomes and enhanced migration ability [24].
3. Damaged mitochondrial DNA is a common byproduct of autophagy and can induce in flammation via NLRP3 activation [20, 21].
4. β-Hydroxybutyrate, a ketone that occurs during starvation, has been shown to reduce in flammasome activation by reducing potassium efflux[25]. Although its effects on atherosclerosis are unknown, it may explain the anti-in flammatory basis of the ketogenic diet.
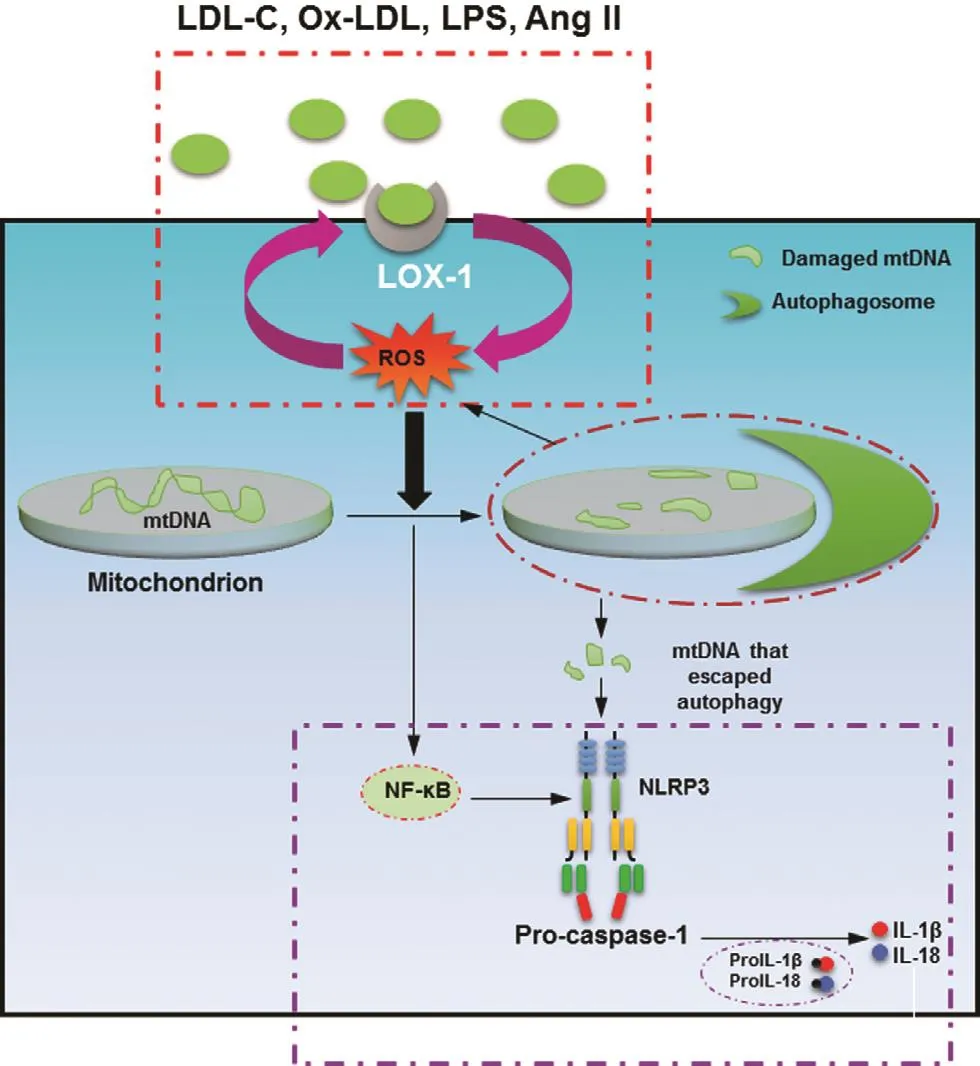
Figure 3 Lectin-Type Oxidized LDL Receptor 1 (LOX1)Mediates In flammasome Activation Through Damaged Mitochondrial DNA.
5. NLRP3 expression may also mediate the atherogenic effects of cellular hypoxia within the atheromatous plaque [26]. Hypoxia is commonly present because of reduced diffusion as well as increased consumption within the plaque.Cellular hypoxia within the plaque promotes foam cell and necrotic core formation as well as catabolism of extracellular matrix by inducing matrix metalloproteinases. In activated human macrophages, Folco et al. [26] showed that moderate hypoxia also increases the expression of NLRP3, caspase 1 and IL-1β.
Finally, the role of the NLRP3 in flammasome may extend beyond its proin flammatory effects. NLRP3 in flammasomes may also alter macrophage function and alter lipid deposition, which may increase their susceptibility to form foam cells. ASC gene(PYCARD) deletion markedly abolished NLRP3 in flammasome activation, attenuated lysosomal lipid deposition, and decreased macrophage migration ability [24].
Evidence Linking the NLRP3 Pathway to Atherosclerosis and Associated Disease States
In the landmark study by Duewell et al. [11], bone marrow was transplanted from either wild-type or NLRP3-knockout mice into LDL receptor-de ficient mice. Despite similar cholesterol levels, the components of NLRP3 in flammasomes, the levels of the cytokines IL-1 and IL-18, and the extent of early atherosclerosis were markedly decreased in mice that received the NLRP3-de ficient bone marrow.Silencing of NLRP3 also suppresses atherosclerosis and stabilizes plaques in ApoE-de ficient mice [27].
There is growing evidence from human studies that supports this mechanism of development of atherosclerosis. NLRP3 in flammasomes are highly expressed in aorta of patients with atherosclerosis undergoing coronary artery bypass grafting and correlate signi ficantly with traditional risk factors such as LDL as well as the severity of coronary artery disease [28]. In a cohort of 123 patients with acute coronary syndrome, peripheral blood monocyte NLRP3 levels were shown to correlate with the severity of coronary atherosclerosis and were a predictor of major adverse cardiac events in a Cox regression model (P=0.043) [29].Components of the NLRP3 pathway are highly expressed in carotid plaque of patients undergoing carotid endarterectomy, with signi ficantly higher levels in unstable plaque compared with stable plaque [30]. Single nucleotide polymorphisms in NLRP3 have been associated with the presence of abdominal aortic aneurysms, another manifestation of atherosclerosis [31].
In flammasomes as Therapeutic Targets
The discovery of in flammasomes thus provides insight into the mechanism that links hyperlipidemia to in flammation, especially the IL-1β pathway. The finding that cholesterol crystals initiate in flammation supports the two major therapeutic strategies already used to combat atherosclerosis: diet and 3-hydroxy-3-methylglutaryl coenzyme A receptor antagonists. Although lipid deposition in the plaque has long been considered a consequence of in flammation and cell death, activation of in flammasomes by cholesterol crystals provides evidence that hyperlipidemia actually promotes in flammation.Intuitively, a healthy diet and lifestyle could help attenuate the effects of in flammasome activation.The effects of dietary interventions on NLRP3 have been studied in adipose tissue but not in human atherosclerosis [32]. Since adipose tissue NLRP3 levels correlate with severity of atherosclerosis [33],this can be considered indirect evidence that dietary modi fication can be helpful. Statins have long been known to attenuate in flammation and reduce the levels of markers such as high- sensitivity C-reactive protein [34]. This was regarded a pleiotropic effect and not necessarily related to their lipid-lowering abilities. Conceptually, lipid lowering with statins could also reduce in flammation by reducing in flammasome activation. Preliminary human data suggest that statins reduce NLRP3 activation [35, 36].Treatment with ezetimibe, a nonstatin drug that works by inhibiting absorption of cholesterol in the intestine, has also been shown to reduce cholesterol crystal formation in rabbits and reduce serum cholesterol levels [37].
The discovery of NLRP3 has provided insight into the anti-in flammatory and antiatherosclerotic mechanism of action of other agents as well.Resveratrol is a flavanoid that has been shown to reduce in flammation; this effect may be mediated by attenuation of the NLRP3 pathway [38]. NLRP3 has also been implicated in the salutary effects of moderate alcohol intake on coronary artery disease[39]. The antiatherosclerotic effect of dipeptidyl dipeptidase 4 inhibitors (antidiabetic drugs) may also be through suppression of NLRP3 via glucagon-like peptide 1 receptor [40].
At least three major components of the in flammasome pathway – NLRP3, caspase 1, and IL-1β, are potential therapeutic targets.
IL-1β Antagonists
IL-1β antagonists are already in clinical use in the treatment of diseases such as gout and rare genetic syndromes such as Muckle-Wells syndrome (an autosomal dominant disorder characterized by mutations in theNLRP3gene resulting in abnormal in flammatory response). The Canakinumab Anti-in flammatory Thrombosis Outcomes Study(CANTOS) is designed to study the efficacy of canakinumab, an antibody that selectively inhibits IL-1β to reduce plaque in flammation and the risk of cardiovascular events [41]. The study will provide indirect evidence of the bene fits of attenuating in flammasome formation. In addition, since canakinumab does not affect lipid parameters, the study will provide proof of the bene fits of targeting in flammation in coronary artery disease [42].
Caspase 1 Inhibition
The antiatherosclerotic effects of caspase 1 inhibition in animal models is less clear. Menu et al. [43]fedApoe−/−/Nlrp3−/−,Apoe−/−/Pycard−/−, andApoe−/−/Casp1−/−double-de ficient mice a high-fat diet for 11 weeks and subsequently assessed atherosclerosis progression and plaque phenotype in comparison withApoe−/−mice. No differences in atherosclerosis progression, in filtration of plaques by macrophages,or plaque stability was found [43]. However, Usui et al. [44] demonstrated reduced vascular in flammation and atherosclerosis in ApoE/caspase 1 double-de ficient mice. Similarly Gage et al. [45]found a reduction in atherosclerosis despite similar lipid levels in caspase 1/ApoE double-de ficient mice compared with ApoE-de ficient mice. The difference in these studies has been attributed to the difference in the composition of the diet [46].
Although caspase 1 is primarily responsible for cleavage of pro-IL-1β intracellularly, other proteases such as neutrophil elastase can extracellularly process the pro-IL forms into active cytokines [47,48]. Caspase 1–de ficient mice have low, but comparable, amounts of circulating active IL-1β compared with wild-type controls after carotid ligation and do not exhibit a statistically signi ficant reduction in neointima formation [14]. They also mount a full in flammatory response to subcutaneous turpentine, another IL-1β-dependent process [49]. Thus the effect of caspase 1 inhibition on atherosclerosis is uncertain.
Although several caspase 1 inhibitor molecules are under preliminary investigation in various diseases, none are in clinical trials in patients with atherosclerosis.
NLRP3 Inhibition
Preliminary animal data suggest that inhibiting NLRP3 itself may be potentially useful. Silencing of NLRP3 suppresses atherosclerosis and stabilizes plaques inApoE-de ficient mice [27]. Arglabin, an anti-in flammasome inhibitor, has been shown to reduce in flammation and atherosclerosis in ApoE-de ficient mice [46]. A novel small molecule NLRP3 inhibitor, MCC950, has been developed but has not been studied yet in atherosclerosis [50]. Various acrylamide derivatives are being developed for their anti-in flammasome activity via inhibition of NLRP3 ATPase [51].
Conclusion
In flammasomes play an important role in innate immunity, and the NLRP3 in flammasome plays a major role in mediating atherosclerosis. Its discovery has improved our understanding of how disorders of cholesterol metabolism contribute to in flammation. Whether targeting in flammasomes directly would be a viable therapeutic target in atherosclerosis remains to be seen.
Conflict of Interest
The authors declare no Conflict of interest.
REFERENCES
1. Libby P, Ridker PM, Maseri A.In flammation and atherosclerosis.Circulation 2002;105(9):1135–43.
2. Martinon F, Burns K, Tschopp J.The in flammasome: a molecular platform triggering activation of in flammatory caspases and processing of proIL-β. Mol Cell 2002;10(2):417–26.
3. Packard RR, Libby P. In flammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem 2008;54(1):24–38.
4. Hansson GK. In flammation,atherosclerosis, and coronary artery disease. N Engl J Med 2005;352(16):1685–95.
5. Stutz A, Golenbock DT, Latz E.In flammasomes: too big to miss. J Clin Invest 2009;119(12):3502–11.
6. Guo H, Callaway JB, Ting JP.In flammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015;21(7):677–87.
7. Latz E, Xiao TS, Stutz A.Activation and regulation of the in flammasomes. Nat Rev Immunol 2013;13(6):397–411.
8. Tsutsui H, Imamura M, Fujimoto J, Nakanishi K. The TLR4/TRIF-mediated activation of NLRP3 in flammasome underlies endotoxin-induced liver injury in mice. Gastroenterol Res Pract 2010;2010:641865.
9. Di Virgilio F. The therapeutic potential of modifying in flammasomes and NOD-like receptors. Pharmacol Rev 2013;65(3):872–905.
10. Pétrilli V, Papin S, Dostert C,Mayor A, Martinon F, Tschopp J. Activation of the NALP3 in flammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007;14:1583–9.
11. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G,Bauernfeind FG, et al. NLRP3 in flammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357–61.
12. Rajama K, Lappalainen J, Oorni K,Valimaki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 in flammasome in human macrophages: a novel link between cholesterol metabolism and in flammation. PLoS One 2010;5:e11765.
13. Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of in flammation. Eur J Immunol 2011;41(5):1203–17.
14. Chamberlain J, Evans D, King A,Dewberry R, Dower S, Crossman D, et al. Interleukin-1β and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol 2006;168:1396–403.
15. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1β decreases the severity of atherosclerosis in apoE-de ficient mice. Arterioscler Thromb Vasc Biol 2003;23:656–60.
16. Badimon L. Interleukin-18: a potent pro-in flammatory cytokine in atherosclerosis. Cardiovasc Res 2012;96(2):172–5.
17. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, et al. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 in flammasome activation.Biochem Biophys Res Commun 2012;425(2):121–6.
18. Sheedy FJ, Grebe A, Rayner KJ,Kalantari P, Ramkhelawon B,Carpenter SB, et al. CD36 coordinates NLRP3 in flammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile in flammation. Nat Immunol 2013;14(8):812–20.
19. Janoudi A, Shamoun FE,Kalavakunta JK, Abela GS.Cholesterol crystal induced arterial in flammation and destabilization of atherosclerotic plaque. Eur Heart J 2016;37(25):1959–67.
20. Ding Z, Liu S, Wang X, Dai Y,Khaidakov M, Deng X, et al.LOX-1, mtDNA damage, and NLRP3 in flammasome activation in macrophages: implications in atherogenesis. Cardiovasc Res 2014;103(4):619–28.
21. Ding Z, Liu S, Wang X, Theus S,Fan Y, Deng X, et al. LOX-1 –dependent mitochondrial DNA damage and NLRP3 activation during systemic in flammation in mice.Biochem Biophys Res Commun 2014;451(4):637–43.
22. Estruch M, Rajamäki K, Sanchez-Quesada JL, Kovanen PT, Öörni K, Benitez S, et al. Electronegative LDL induces priming and in flammasome activation leading to IL-1β release in human monocytes and macrophages. Biochim Biophys Acta 2015;1851(11):1442–9.
23. Oury C. CD36: linking lipids to the NLRP3 in flammasome, atherogenesis and atherothrombosis. Cell Mol Immunol 2014;11(1):8–10.
24. Li X, Zhang Y, Xia M, Gulbins E,Boini KM, Li P-L. Activation of NLRP3 in flammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of in flammasome in atherogenesis. PLoS One 2014;9(1):e87552.
25. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M,Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 in flammasome-mediated in flammatory disease. Nat Med 2015;21(3):263–9.
26. Folco EJ, Sukhova GK, Quillard T,Libby P. Moderate hypoxia potentiates interleukin-1β production in activated human macrophages.Circ Res 2014;115(10):875–83.
27. Zheng F, Xing S, Gong Z, Mu W,Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-de ficient mice. Mediat In flamm 2014;2014:507208.
28. Zheng F, Xing S, Gong Z, Xing Q.NLRP3 in flammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ 2013;22:746–50.
29. Afrasyab A, Qu P, Zhao Y, Peng K,Wang H, Lou D, et al. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients.Heart Vessels 2016;31(8):1218–29.
30. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 in flammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis 2015;24(11):2455–66.
31. Roberts RL, Van Rij AM, Phillips LV,Young S, McCormick SP, Merriman TR, et al. Interaction of the in flammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms.Atherosclerosis 2011;218(1):123–6.
32. Finucane OM, Lyons CL, Murphy AM, Reynolds CM, Klinger R,Healy NP, et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 in flammasome-mediated IL-1β secretion and insulin resistance despite obesity.Diabetes 2015;64:2116–28.
33. Bando S, Fukuda D, Soeki T,Nishimoto S, Uematsu E, Matsuura T, et al. Expression of NLRP3 in subcutaneous adipose tissue is associated with coronary atherosclerosis.Atherosclerosis 2015;242:407–14.
34. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial.Lancet 2009;373(9670):1175–82.
35. Altaf A, Qu P, Zhao Y, Wang H,Lou D, Niu N. NLRP3 in flammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron Artery Dis 2015;26(5):409–21.
36. Satoh M, Tabuchi T, Itoh T,Nakamura M. NLRP3 in flammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci (Lond)2014;126(3):233–41.
37. Patel R, Janoudi A, Vedre A, Aziz K, Tamhane U, Rubinstein J, et al.Plaque rupture and thrombosis is reduced by lowering cholesterol levels and crystallization with ezetimibe and is correlated with FDG-PET. Arterioscler Thromb Vasc Biol 2011;31:2007–14.
38. Deng ZY, Hu MM, Xin YF, Gang C. Resveratrol alleviates vascular in flammatory injury by inhibiting in flammasome activation in rats with hypercholesterolemia and vitamin D2 treatment. In flamm Res 2015;64(5):321–32.
39. Nurmi K, Virkanen J, Rajamäki K, Niemi K, Kovanen PT, Eklund KK. Ethanol inhibits activation of NLRP3 and AIM2 in flammasomes in human macrophages–a novel anti-in flammatory action of alcohol. PLoS One 2013;8:e78537.
40. Dai Y, Dai D, Wang X, Ding Z,Mehta JL. DPP-4 inhibitors repress NLRP3 in flammasome and interleukin-1beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc Drugs Ther 2014;28:425–32.
41. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-in flammatory Thrombosis Outcomes Study (CANTOS). Am Heart J 2011;162(4):597–605.
42. Ridker PM, Howard CP, Walter V, Everett B, Libby P, Hensen J,et al. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation 2012;126(23):2739–48.
43. Menu P, Pellegrin M, Aubert JF,Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-de ficient mice progresses independently of the NLRP3 in flammasome.Cell Death Dis 2011;2:e137.
44. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A,Karasawa T, et al. Critical role of caspase-1 in vascular in flammation and development of atherosclerosis in Western diet-fed apolipoprotein E-de ficient mice.Biochem Biophys Res Commun 2012;425(2):162–8.
45. Gage J, Hasu M, Thabet M,Whitman SC. Caspase-1 de fi-ciency decreases atherosclerosis in apolipoprotein E-null mice. Can J Cardiol 2012;28:222–9.
46. Abderrazak A, Couchie D,Mahmood DF, Elhage R, Vindis C, Laffargue M, et al. Antiin flammatory and antiatherogenic effects of the NLRP3 in flammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 2015;131:1061–70.
47. Hazuda DJ, Strickler J, Kueppers F,Simon PL, Young PR. Processing of precursor interleukin 1 beta and in flammatory disease. J Biol Chem 1990;265:6318–22.
48. Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V,Chamberlain J, et al. Neutrophil elastase promotes interleukin-1β secretion from human coronary endothelium. J Biol Chem 2015;290(40):24067–78.
49. Fantuzzi G, Ku G, Harding MW,Livingston DJ, Sipe JD, Kuida K,et al. Response to local in flammation of IL-1 beta-converting enzyme-de ficient mice. J Immunol 1997;158:1818–24.
50. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A smallmolecule inhibitor of the NLRP3 in flammasome for the treatment of in flammatory diseases. Nat Med 2015;21:248–55.
51. Cocco M, Miglio G, Giorgis M,Garella D, Marini E, Costale A, et al. Design, synthesis, and evaluation of acrylamide derivatives as direct NLRP3 in flammasome inhibitors. Chem Med Chem 2016;11(16):1790–813.
 Cardiovascular Innovations and Applications2016年3期
Cardiovascular Innovations and Applications2016年3期
- Cardiovascular Innovations and Applications的其它文章
- Outcome Trials in the Therapeutic Management of Hypertension in East Asians
- The Gut Microbiota and Atherosclerosis:The State of the Art and Novel Perspectives
- Psychosocial Risk Factors and Cardiovascular Disease: Epidemiology, Screening, and Treatment Considerations
- Management of Hypertension: JNC 8 and Beyond
- ACC/AHA Guidelines for Cardiovascular Disease Prevention and Cholesterol Management:Implications of New Therapeutic Agentsa
- Smoking and Passive Smoking