
膜荚黄芪的转录组测序质量评估及其SSR位点信息分析的研究
2016-05-14 11:06常越闫嵩刘振鹏任伟超刘玠马伟
中国中药杂志 2016年8期
常越 闫嵩 刘振鹏 任伟超 刘玠 马伟

[摘要]该实验采用Roche 454 GS FLX测序仪获得黄芪的转录组数据,使用454 Sequencing System Software分析软件进行转录组从头拼接;利用MISA工具筛选了黄芪转录组测序获得的9 893条unigenes,对其SSR 位点信息进行了分析。结果表明,进行测序所得的reads的平均长度为413 bp,约86%的reads参与了拼接,拼接的N50长度为1 205 bp,所测得的unigene数量基本涵盖了全部转录组信息;黄芪转录组搜索到1 729个SSR位点,SSR的发生频率为924%,SSR在黄芪整个转录组中出现的频率为1342%,SSR的平均距离为797 kb。一共发现核心重复序列127种,占优势的是二核苷酸型中的TG/AC型,出现的频率占总SSR位点的425%。黄芪转录组的测序结果揭示了黄芪转录组的整体表达特征,并得到大量黄芪转录组unigene序列,并且黄芪转录组SSR位点出现频率高,类型多样,多态性潜能高。
[关键词]膜荚黄芪;转录组测序;SSR;位点信息
[Abstract]In this study, 454/Roche GS FLX sequencing technology was used to obtain the data of the Astragalus membranaceus Four hundred and fiftyfour Sequencing System Software was applied to carry out the transcription of the group from scratch Using MISA tools, 9 893 unigenes were selected for the sequence of the genome of A membranaceus, and the information of SSR locus was analyzed According to the result, the average length of reads was 413 bp, about 86% of the reads was involved in the splicing, the length of the N50 was 1 205 bp, the number of unigenes was measured by the whole transcript 1 729 SSR loci in the A membranaceus transcriptome were searched, the occurrence frequency of SSR was 924%, the frequency of SSR in the whole transcriptome was 1342%, the average length of SSR was 797 kbOne hundred and twentyseven kinds of core repeat sequences were found, the dominant type was TG/AC type of dinucleotide, it appeared to account for 425% of the total SSR locus The results of the sequence of the transcription of the A membranaceus transcriptome revealed the overall expression, and a large number of unigenessequence was obtained, and the SSR locus in the genome of the A membranaceus is high, and the type is diverse, and the polymorphism of the gene is high
[Key words]Astragalus membranaceus; transcriptome sequencing; SSR; information of loci
doi:10.4268/cjcmm20160810
中药材黄芪按《中国药典》(2010年版)规定,是蒙古黄芪Astragalus membranaceus(Fisch) Bgevarmongholicus Hsiao Bge或膜荚黄芪A membranaceus(Fisch) Bge的干燥根。味甘性温,具有补气升阳,健脾利尿,排毒排脓,敛疮生肌等功效[1]。转录组(transcriptome)是指特定细胞在某一功能状态下全部表达的基因总和,代表了每一个基因的身份和表达水平,转录组测序能全面的地揭示生物个体在特定组织和特定时期的全局基因的表达情况。简单重复序列(simple sequence repeats,SSR),一般以1~6个碱基为核心序列,具有高度多态性。目前已利用SSR 标记构建了许多物种的染色体遗传图谱,并被广泛应用于基因定位及亲缘关系分析、品种鉴定和动植物育种等领域[2]。本实验旨在完成黄芪转录组测序并分析其SSR位点信息,为黄芪进一步在遗传图谱构建、种质鉴定、遗传多样性分析等方面的研究奠定了基础。
1材料与方法
11植物
本试验所用膜荚黄芪种子,由黑龙江中医药大学试验中心马伟研究员鉴定。在黑龙江中医药大学药用植物园日光温室内无土栽培培养,选取苗龄40 d的黄芪。取材健康的根、茎、叶,采集后用液氮速冻后-80 ℃保存。
12植物总RNA的提取
黄芪总RNA提取参考改良的CTAB方法[3],具体步骤如下:取适量植物材料于研钵中加入液氮研磨至粉末状;将粉末转移至20 mL离心管中,加入1 mL无水乙醇,充分振荡后,冰上放置10 min,4 ℃ 12 000 r·min-1离心2~5 min;小心弃掉上清,在离心管中加入900 μL CTAB提取液,振荡混匀后,65 ℃水浴5 min,放置冰上,加入等体积氯仿,4 ℃ 12 000 r·min-1离心5~10 min;小心吸取上清至新的离心管中,加入1/2体积无水乙醇及08体积5 mol·L-1LiCl,混匀,冰上放置10 min,4 ℃ 12 000 r·min-1离心10 min,弃上清;70%乙醇洗涤沉淀2次,室温放置5 min干燥RNA,加入50 μL灭菌水溶解RNA,-80 ℃备用。
13mRNA 样品准备
131总RNA的质量检测与定量使用紫外分光光度计,对总RNA的浓度、总量及A260/280和A260/230进行分析。采用Agilent RNA 6000 Pico Kit;Agilent,50671513,对总RNA的完整性、纯度及降解度进行测定。
132mRNA的纯化与定量采用Oligotex mRNA Mini Kit;Qiagen, 70022;Agilent RNA 6000 Pico Kit;Agilent,50671513试剂盒,进行mRNA 纯化与定量。
14cDNA文库制备
mRNA片段化至主峰在450~1 200 nt;以1∶1∶1混合根、茎、叶总RNA;采用DNA Synthesis system,Roche,11117831001和AgencourtAMPure XP Beads,Beckman,A63881试剂盒,进行双链 cDNA 合成与纯化;采用Roche Rapid Library Preparation Kit,Roche,5608228001试剂盒,进行片段末端修复;连接接头;采用AgencourtAMPure XP Beads,Beckman,A63881试剂盒,去除小片段;使用仪器QuantifluorST fluorometer,Promega,E6090,进行cDNA 文库定量;使用仪器Agilent 2100 Bioanalyzer,Agilent,2100和Agilent High Sensitivity DNA Kit,Agilent,50674626试剂盒,进行cDNA文库质量检测;准备工作液。
15emPCR扩增
通过乳液滴定或测序滴定确定emPCR扩增中所需的DNA文库的量;准备emPCR各试剂、乳化油、Mock Amplification Mix 和预乳液、Live Amplification Mix;使用DNA Capture Beads将DNA文库捕获;乳化;扩增;回收DNA Capture Beads;含DNA文库的DNA Capture Beads的富集;测序引物退火。
16Roche 454 GS FLX+测序仪上机测序
转录组测序工作委托上海派森诺生物科技有限公司完成。
17原始数据整理、过滤及质量评估
下机数据经过454Newbler去接头、去低质量碱基处理。使用454 Sequencing System Software (http://www454com/)分析软件进行转录组从头拼接[45]。
18测序饱和度分析
测序饱和度是随着采样读取的测序量(sampled reads number)的增加,而检测到的unigene数目的变化情况。当采样读取的测序量到一定程度,而检测到的unigene几乎不增加或者很少增加,则测序包和,否者就是测序量不够没有达到饱和。在本次测序数据中,以15 000作为梯度,对数据依次随机抽样,看抽出来的这些reads分别检测到多少unigene。然后把采样读取的测序量做横坐标和检测到的unigene的数量做纵坐标画一个曲线,查看这条曲线随着采样读取数据量的增加unigene是否具有饱和性。
19转录组SSR位点的信息分析
将转录组数据用MISAMIcroSAtellite identification tool( http://pgrcipkgaterslebende/misa/ )进行SSR分析。程序配置为:1/10,2/6,3/5,4/5,5/5,6/5(串联重复的核心序列/最小重复单位数目);复合型SSR中间隔串联核心序列的非重复碱基数最大不超过100[68]。
2结果与分析
21总RNA质量分析
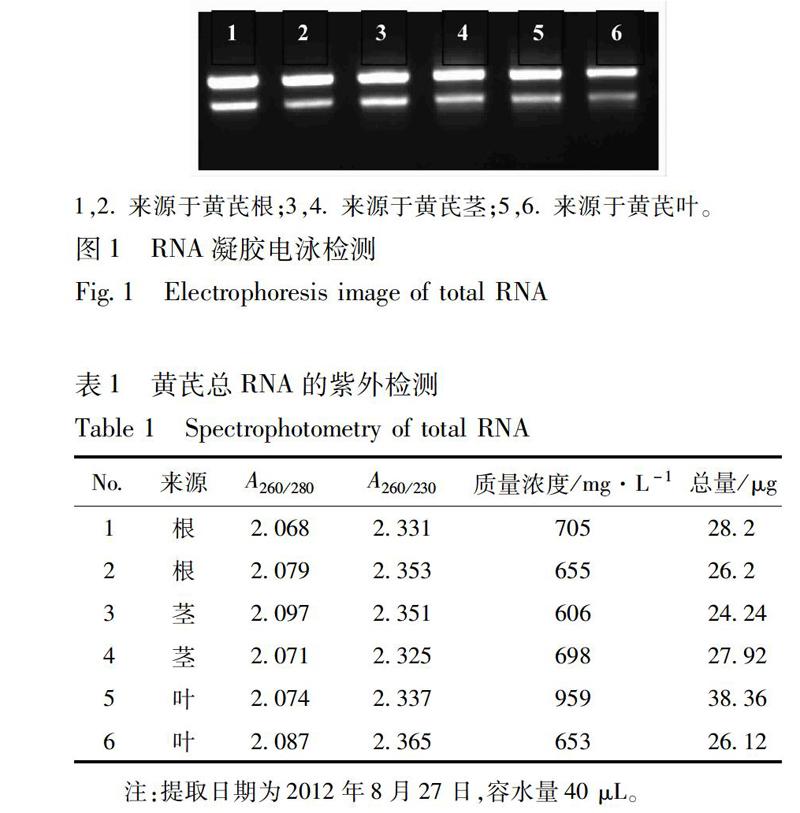
质量浓度≥250 mg·L-1,总量≥50 μg;A260/28018~22,A260/230应≥20。电泳检测28S∶18S至少大于15,见图1;RIN≥80;并确保RNA无降解,无污染,见表1。
22测序量统计
测得原始数据量约513 Mb,通过过滤得到符合拼接要求的有效数据306 Mb。数据总汇后,数据量为306 805 437,基因条数为742 721条,平均长度为413 kb;基因统计后,基因为742 721条,读长大于20 bp序列数为742 602,原始数据为513 365 449,过滤后数据为306 805 437,拼接利用率为5976%。
经过转录组从头拼接,8604%的reads参与拼接,共计639 061条reads。序列拼接的N50长度为1 205 bp,长度>N50为3 823 bp,平均长度为1 0695 bp。拼接得到contig 15 167个,这些contig继而拼接成为12 851个isotig。无法进一步拼接成为isotig的contig与这些isotig一起,组成10 742个isogroup,总共得到12 880条Isotig(包括无法进一步拼接成为isotig的contig),Unigene数量为9 893,Unigene平均长度为1 097128 98 bp。
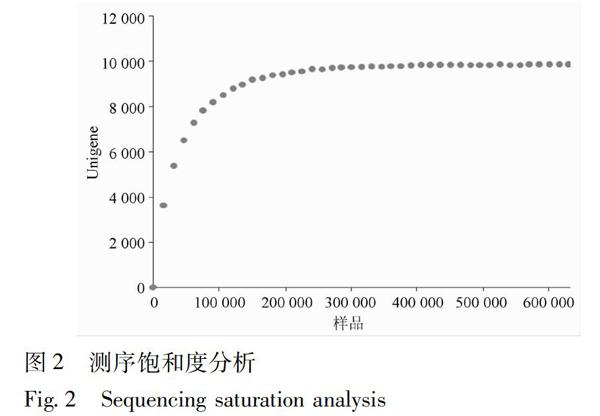
23测序饱和度分析
当采样读取测序量达到2×102 kb时,unigene的数目已趋于饱和,当采样读取数据量达到3×102 kb unigene的数据已经完全饱和。这说明对于本次黄芪根、茎、叶的等比例混样测序,测序量已经达到饱和,测序量满足试验要求,见图2。
24SSR位点的数量与分布
对黄芪转录组的12 880条isotig进行SSR的搜索,得到SSR的总碱基数是23 834 bp,共找到SSR位点1 729个,复合型SSR位点435个,含有SSR位点的isotig条数为1 190条,包含一个以上SSR位点的isotig的条数是252条。SSR的发生频率(含有SSR的unigene数目与总unigene数目的比值)924%;SSR在黄芪整个转录组中出现的频率(SSR位点个数和总unigene数目的比值)1342%;黄芪转录组中SSR的平均距离(总unigene的长度与SSR数目的比值)797 kb,见表2。
从表2可以看出黄芪转录组SSR种类丰富,从单核苷酸到六核苷酸的各种核酸重复类型都能够看到,但他们相差的比列较大。SSR类型多集中在单核苷酸、二核苷酸、三核苷酸上,占963%,其他类型相对的占有量很低。
黄芪转录组SSR位点序列总长度达到23 834 bp,其中三核苷酸核心重复序列的总长度最长,为11 433 bp;其次是二核苷酸,为4 472 bp;接下来是单核苷酸,是1 919 bp。SSR位点的平均长度是24 bp,各类型SSR位点的平均长度分别是12,18,18,24,27,32 bp。
25SSR的特性
在黄芪转录组的1 729个SSR位点中共发现核心重复序列127种,其中单核苷酸型4种;二核苷酸型11种;三核苷酸型57种;四核苷酸型28种;五核苷酸型11种;六核苷酸型16种。这6种重复序列类型的重复次数大多集中在5~10次,达到1 173次,占整个重复次数的6784%;其次是10~15次,重复次数是356次,占整个重复次数的2059;第三的是16~20次,重复次数是149次,占整个重复次数的862%。也就是说,核心序列重复次数在5~20次的SSR的出现次数为1 678次,占整个重复次数的9705%,核心序列重复次数超过20次的SSR出现的次数比例还不到3%,见图3。从出现的具体类型分析,占优势的是二核苷酸型中的TG/AC型,出现的频率占总SSR位点的425%;其次是GA/CT型,出现的频率占总SSR位点的394%;接下来是GAA/CTT型,频率是356%;CCT/GGA型,频率是309%,见表3。
26SSR的可用性评价
判断SSR可用性的重要依据是SSR分子标记的多态性,一般认为SSR的长度是影响其多态性高低的重要因素,当SSR≥20 bp时,多态性较高;当12 bp≤SSR≤20 bp时,多态性中等;当SSR≤12 bp时,多态性极低。黄芪转录组数据显示SSR的长度多集中在12~45 bp,其中12 bp≤SSR≤20 bp具有中度多态性的SSR共有861个,占总SSR的4980%;SSR≥20 bp具有高多态性的SSR共422个,占总SSR的2441%。同时有研究表明,高级基元SSR的多态性比低级基元的多态性普遍偏低。在黄芪转录组数据统计分析中发现,长度大于20 bp的SSR大多属于低级基元,共包含SSR位点282个。可以预计这部分多态性潜能高的SSR在黄芪基因功能研究上具有较高的利用价值。
3讨论与结论
31黄芪转录组测序质量评估
黄芪转录组测序所用的根、茎、叶提取的总RNA检测结果合格。进行测序所得的reads的平均长度为413 bp,约86%的reads参与了拼接,拼接的N50长度为1 205 bp。测序饱和度结果表明,所测得的unigene数量基本涵盖了全部转录组信息。从以上结果可知本转录组的测序结果真实可信,可以对数据进行进一步分析。
32黄芪转录组SSR位点信息分析
SSR广泛分布于各种真核生物的基因组中,大约每隔10~50 kb就存在1个SSR。在植物中,平均233 kb就有1个SSR;双子叶植物中的SSR数量大于单子叶植物,前者2个SSR之间的平均间距为212 kb,后者为646 kb。通过对黄芪转录组数据的SSR分析,黄芪转录组中SSR的平均距离是797 kb,从单核苷酸类型到六核苷酸类型均具备,核心重复序列127种,这表明黄芪基因组内具有较高丰度的SSR。
大部分植物的SSR重复基元主要以二、三核苷酸型为主,但不同物种之间的主导SSR重复类型有所差异。本研究发现膜荚黄芪转录组SSR重复类型主要以三核苷酸为主,占全部SSR的4222%,一核苷酸所占比例也较高,占全部SSR的3777%。这与大豆、棉花、大麦、玉米、水稻等主要经济作物的研究结果相同,这些植物也是以三核苷酸重复类型为主[910]。在膜荚黄芪的SSR中的二核苷酸基元中TG/AC类型最多,三核苷酸基元中GAA/CTT类型最多。这些重复类型与人参[11]、丹参[12]、番红花[13]等植物中二元碱基以AG,TC,CT为主要类型不同,在三碱基重复基元中以AAG/TTC,GAA/TCC为主与黄芪相同。据此推断这种重复基元的差别可能与物种差异有关联。从SSR的类型来看,黄芪转录组SSR类型多样。这些类型多样,数量丰富的SSR为黄芪进一步在遗传图谱构建、种质鉴定、遗传多样性分析、标记辅助选择(MAS,marker assistant seletion,marker aided seletion)、基因定位、数量性状基因座(QTL)分析,系谱分析和亲源关系鉴定等方面的研究奠定了基础。
[参考文献]
[1]中国药典. 一部[S]. 2010:11.
[2]闫秋良. 基于生物信息学方法从牛和绵羊表达序列标签中筛选SSR标记的初步研究[D]. 杨凌:西北农林科技大学, 2007.
[3]陈肃,刘雪梅,李发兵. 一种快捷有效的提取树木RNA方法[J]. 辽宁林业科技,2008(5):25
[4]Margulies M, Egholm M, AltmanW E, et al. Genome sequencing in microfabricated highdensity picolitre reactors[J]. Nature,2005,437(7057):376.
[5]Kumar S, Blaxter M L. Comparing de novo assemblers for 454 transcriptome data[J]. BMC Genomics,2010,11(2):237.
[6]Argout X, Fouet O, Wincker P, et al. Towards the understanding of the cocoa transcriptome:production and analysis of an exhaustive dataset of ESTs of Theobroma cacao L. generated from various tissues and under various conditions[J]. BMC Genomics,2008,9(11):512.
[7]Luro F L, Costantino G,Terol J, et al. Transferability of the ESTSSRs developed on Nules clementine(Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping[J]. BMC Genomics,2008,9(12):287.
[8]Simbaqueba J,Sanchez P,Sanchez E,et al. Development and characterization of microsatellite markers for the cape gooseberry physalisperuviana[J]. PLoS ONE,2011,6(10):e26719.
[9]Cardle L, Ramsay L, Milbourne D, et al. Computational and experimental characterization of physically clustered simple sequence repeats in plants[J]. Genetics. 2000,156(2):847.
[10]Varshney R K, Graner A, Sorrells M E. Genic microsatellite markers in plants: features and applications[J]. Trends Biotechnol,2005,23(1):48.
[11]Li C, Zhu Y, Guo X, et al. Transcriptome analysis reveals ginsenosides biosynthetic genes,microRNAs and simple sequence repeats in Panax ginseng C. A. Meyer[J]. BMC Genomics,2013,14:245.
[12]王学勇,周晓丽,高伟,等. 丹参新的ESTSSR分布规律及分子标记的建立[J]. 中国中药杂志,2011, 36 (3):289.
[13]陈国庆. 番红花EST资源的SSR信息分析[J]. 广西植物,2011,31(1):43.
[责任编辑吕冬梅]
