
高通量测序分析人工养殖成年林麝粪便古菌菌群多样性
2016-05-14 07:00王立志徐谊英蔡永华
动物营养学报 2016年2期
王立志 徐谊英 蔡永华
(1.四川农业大学动物营养研究所,动物抗病营养教育部重点实验室,雅安625014;
2.四川养麝研究所,都江堰611845)
高通量测序分析人工养殖成年林麝粪便古菌菌群多样性
王立志1徐谊英1蔡永华2
(1.四川农业大学动物营养研究所,动物抗病营养教育部重点实验室,雅安625014;
2.四川养麝研究所,都江堰611845)
摘要:本试验的目的是采用高通量测序技术研究人工养殖成年林麝粪便中古菌的结构和组成,并对比不同性别之间的差异。选取12只3岁健康的林麝依据性别分为雄性组(M组)和雌性组(F组),每组各6只。采集其新鲜粪便,提取DNA,用古菌通用引物PCR扩增古菌16S rRNA的V4~V5区,用MiSeq 300PE测序平台对扩增产物进行高通量测序,用QIIME等软件对测序序列进行分析统计。结果表明:在从门到属的各级分类水平上,F组与M组古菌的相对丰度差异均不显著(P>0.05)。M组和F组的组内遗传距离分别为0.16±0.03和0.27±0.06,组间为0.24±0.07,样品的相似度很高。林麝粪便中的古菌可以分为3个门,优势门为阔古细菌门(Euryarchaeota);在属水平上可以分为7个已知属,优势属为甲烷短杆菌属(Methanobrevibacter),其次为热裸单胞菌属(Thermogymnomonas)。结果提示,雄性和雌性林麝粪便中古菌的结构和组成都没有显著差异,Methanobrevibacter是林麝粪便中的优势古菌属
关键词:林麝;古菌;产甲烷古菌;多样性
林麝(Moschusberezovskii)属偶蹄目,麝科,麝属,是一种珍贵的野生药用、香料资源动物,曾经广泛分布于我国的中部和南部[1]。近年来由于栖息地遭受破坏和过度捕猎,数量已急剧下降。目前已被国际自然和自然资源保护联盟(IUCN)列为濒危物种(http://www.iucnredlist.org/details/13894/0)。
我国从20世纪60年代开始人工驯化养殖林麝,至今已有近60年的历史,但发展速度很慢。其中一个主要的原因是人们对林麝的消化生理知识了解很少。林麝是草食性反刍动物,但其胃肠道微生物菌群的组成和结构还不为人所知。为了更好保护和人工驯化养殖,有必要对林麝胃肠道微生物菌群进行系统研究。众所周知,反刍动物瘤胃中栖息着大量微生物,包括细菌、真菌、原虫和古菌等。近年来瘤胃产甲烷古菌越来越受到人们的关注,因为人们相信它们与全球越来越严重的温室效应有关[2]。产甲烷古菌广泛存在于草食动物的胃肠道中,迄今为止,人们已经从水牛[3-4]、绵羊[5]、袋鼠[6]、奶牛[7]、犀牛[8]、肉牛[9-10]、骆驼[11]、牦牛[12]等动物上分离鉴定出了多种产甲烷古菌。综合这些研究可以发现,动物胃肠道产甲烷古菌的多样性具有宿主特异性。因此作者推测,相对于其他动物而言,林麝肠道古菌的结构和组成可能有很大的差异。而有关林麝肠道产甲烷古菌的研究还未见报道。本试验的目的就是用高通量测序技术对林麝粪便中古菌的结构和组成进行研究,并对比不同性别之间的差异。
1材料与方法
1.1试验动物及样品采集
本次试验动物为12只人工养殖的未用过抗生素的健康林麝,根据其性别分为雄性组(M组)和雌性组(F组)2组,每组各6只(6只雄性林麝编号为M1、M2、M3、M4、M5、M6;6只雌性林麝编号为F1、F2、F3、F4、F5、F6),年龄均为3岁,平均体重为(7.58±1.60) kg,饲养于四川省养麝研究所(中国四川都江堰)。林麝单笼饲养,每天饲喂2次,时间为08:00和17:00,自由饮水。饲粮组成及营养水平见表1,在采样之前已稳定饲喂该饲粮28 d以上。于第29天清晨饲喂前戴上消毒过的无菌一次性手套用干净竹签采集动物自然排出的粪便并从粪便深处采集,每头林麝收集新鲜粪便20 g,液氮保存后带回实验室,保存于-80℃冰箱,用于提取DNA。
1.2DNA抽提
用TIANamp微生物DNA Kit[天根生化科技(北京)有限公司]提取林麝粪便样品中微生物总DNA,具体提取步骤参照试剂盒说明书进行。用DNA纯化试剂盒对提取的DNA进行纯化,然后用原子分光光度计和琼脂糖凝胶电泳检验总DNA浓度和纯度后于-20 ℃保存备用。
1.316S rRNA基因扩增及测序
以林麝粪便微生物总DNA为模板,采用古菌通用引物对:341F(5′-CCTAYGGGRBGCASCAG-3′)和806R(5′-GGACTACHVGGGTWTCAAT-3′)[13],PCR扩增古菌16S rRNA的V4~V5区。采用50 μL扩增体系:上、下游引物各0.5 μL,2.5 mmol/L dNTP混合物5 μL,5 μL的10×Ex Taq buffer(含20 mmol/L Mg2+,TaKaRa公司),0.25 μL的Ex Taq DNA polymerase(TaKaRa公司),1 μL的样品DNA模板和37.75 μL的milli-Q水。PCR反应条件为:94 ℃变性3 min;94 ℃ 30 s,50 ℃ 30 s,72 ℃ 30 s,进行30个循环;72 ℃,延伸5 min。每个样品进行3个重复,将同一样品的PCR产物混合后用2%琼脂糖凝胶电泳检测,用QIAquick凝胶回收试剂盒(QIAGEN公司)切胶回收古菌PCR产物,并用试剂盒(QIAGEN公司)进行纯化。纯化的PCR产物送Macrogen生物技术公司(韩国),用Illumina公司的MiSeq 300PE测序平台进行高通量测序。

表1 饲粮组成及营养水平(干物质基础)
1)复合维生素为每千克饲粮提供 Multi-vitamin provided the following per kg of the diet:VA 3 000 IU,VD 350 IU,VE 30 IU。
2)复合微量矿物元素为每千克饲粮提供 Multi-mineral provided the following per kg of the diet:Fe (as ferrous sulfate) 30 mg,Cu(as copper sulfate) 10 mg,Zn (as zinc sulfate) 50 mg,Mn (as manganese sulfate) 60 mg。
3)计算值 Calculated values。
1.4数据分析
使用FLASH软件将测序所得的测序序列(reads)进行双端融合(merge),最小重叠区域为10 bp,最大重叠区域为220 bp。成功merge的序列,使用QIIME软件[14]按照条码(barcode)序列区分各个样品,去除低质量序列。标准如下:去除所有含有N的reads;去除含有连续3个小于Q20碱基的reads;去除与barcode序列不一致的reads;去除连续Q20碱基长度小于序列总长75%的reads。将barcode切除掉后,根据相似性≥97%原则将通过质控的有效序列聚类成为运算分类单位(operational taxonomic unit,OTU),并挑选丰度最高的序列作为OTU的代表性序列;使用QIIME软件将OTU代表序列与RDP数据库(The Ribosomal Database Project,https://rdp.cme.msu.edu/)进行比对,参数为默认参数(相似度为0.9),比对完毕后使用perl脚本去除所有非古菌OTU。用QIIME软件剔除嵌合体和Singletons,并在界、门、纲、目、科及属水平上进行物种注释。采用QIIME默认参数计算各个样品alpha多样性指数[Shannon指数、ChaoⅠ指数、观察物种(observed species)指数]。根据各个样品OTU的种类及其丰度,计算样品间的遗传距离,用非加权UniFrac距离(unweighted UniFrac distance)表示,根据计算结果采用非加权配对算术平均法(unweighted pair group method with arithmetic mean,UPGMA)对样品进行聚类分析。利用perl脚本挑选所有样本中共同存在的共享属,用R软件绘制热图。2个处理间数据的差异性采用SPSS 19.0统计软件进行t检验。
2结果
2.1序列及OTU统计
将测序得到的双端序列首先根据序列之间的重叠关系,将成对的reads拼接成1条序列,经过质控之后,最终获得13 130条属于古菌的有效序列,平均每个样品(1 094±164)条。基于相似度大于97%基础上,将获得的有效序列进行OTU聚类,共获得720个属于古菌的OTU,其中M组385个[平均值为(80.3±24.5)个],F组409个[平均值为(78.8±27.7)个],组间共享OTU为74个(图1)。经计算,M组和F组的序列覆盖度(coverage)分别为80.78%和81.91%。
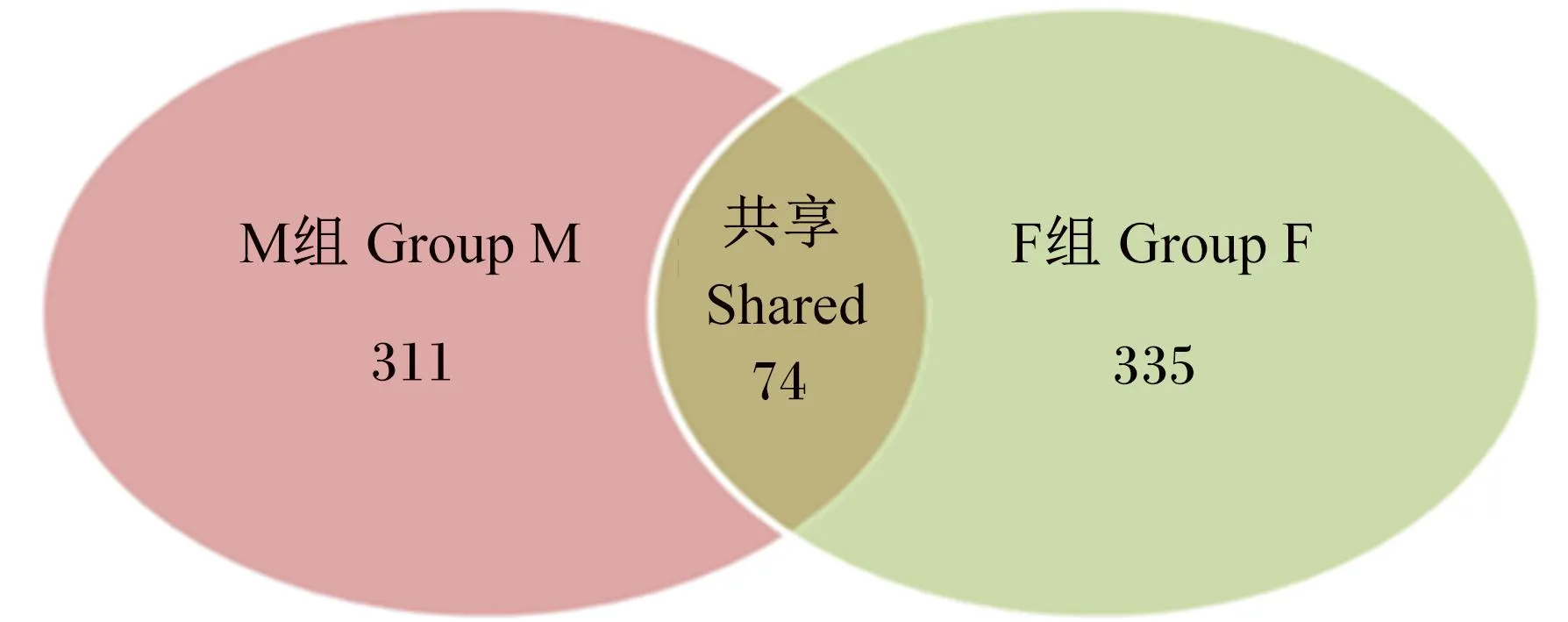
图1 雄性林麝组和雌性林麝组粪样古菌OUT维恩图
2.2粪便物种组成分析
将获得的OTU分别从门到属水平上进行物种注释,具体结果见表2。可以看出,本次试验所得到的90%以上OTU都属于阔古细菌门(Euryarchaeota),而圆齿古细菌门(Crenarchaeota)和未分类门的相对丰度较低。2组间在各分类水平上的相对丰度差异均不显著(P>0.05)。Crenarchaeota的古菌全部来源于热变形菌纲(Thermoprotei),它可进一步分为2个目。其中只有Fervidicoccales可进一步分为1个科和1个属。Euryarchaeota由甲烷菌纲(Methanobacteria)、甲烷微菌纲(Methanomicrobia)、热原体纲(Thermoplasmata)和未分类纲4个纲组成。其中未分类纲不能进一步进行分类,其他3个纲共可分为4个目、6个科、7个属。Methanobacteria由1个目、1个科、3个属组成;Methanomicrobia由2个目、3个科、3个属组成;Thermoplasmata由1个目、2个科组成,在科水平上只有未确定的热源体科(Thermoplasmatales incertae sed)能进一步分到属。在属水平上,相对丰度平均值较高的是甲烷短杆菌属(Methanobrevibacter),其次为热源体属(Thermogymnomonas)。虽然在属水平上2组的差异均不显著(P>0.05),但与M组相比,F组Fervidicoccus相对丰度有降低的趋势(P=0.10)。
2.3alpha多样性分析
表3列出了M组和F组样品的Shannon指数、ChaoⅠ指数、observed species指数。alpha多样性指数主要用于评价样品中微生物的丰富性和均匀性。可以看出,M组的alpha多样性指数在数值上都大于F组,但差异不显著(P>0.05)。
2.4beta多样性分析
根据各个样品OTU的种类及其丰度计算了各个样品间的遗传距离,值越小表示样品间微生物群落结构越接近,相反,值越大则表示样品间微生物群落结构差异越大。M组和F组各自的遗传距离分别为0.16±0.03和0.27±0.06,2组间遗传距离为0.24±0.07,基于此,对样品进行了聚类分析并绘制了PCoA聚类图(图2)。可以看出,除了样品M4和样品M2外,其他10个样品聚集度非常高。虽然F组较M组的聚集程度高,但2组样品在图上并没有依据其组别而表现出明显的组内聚集。
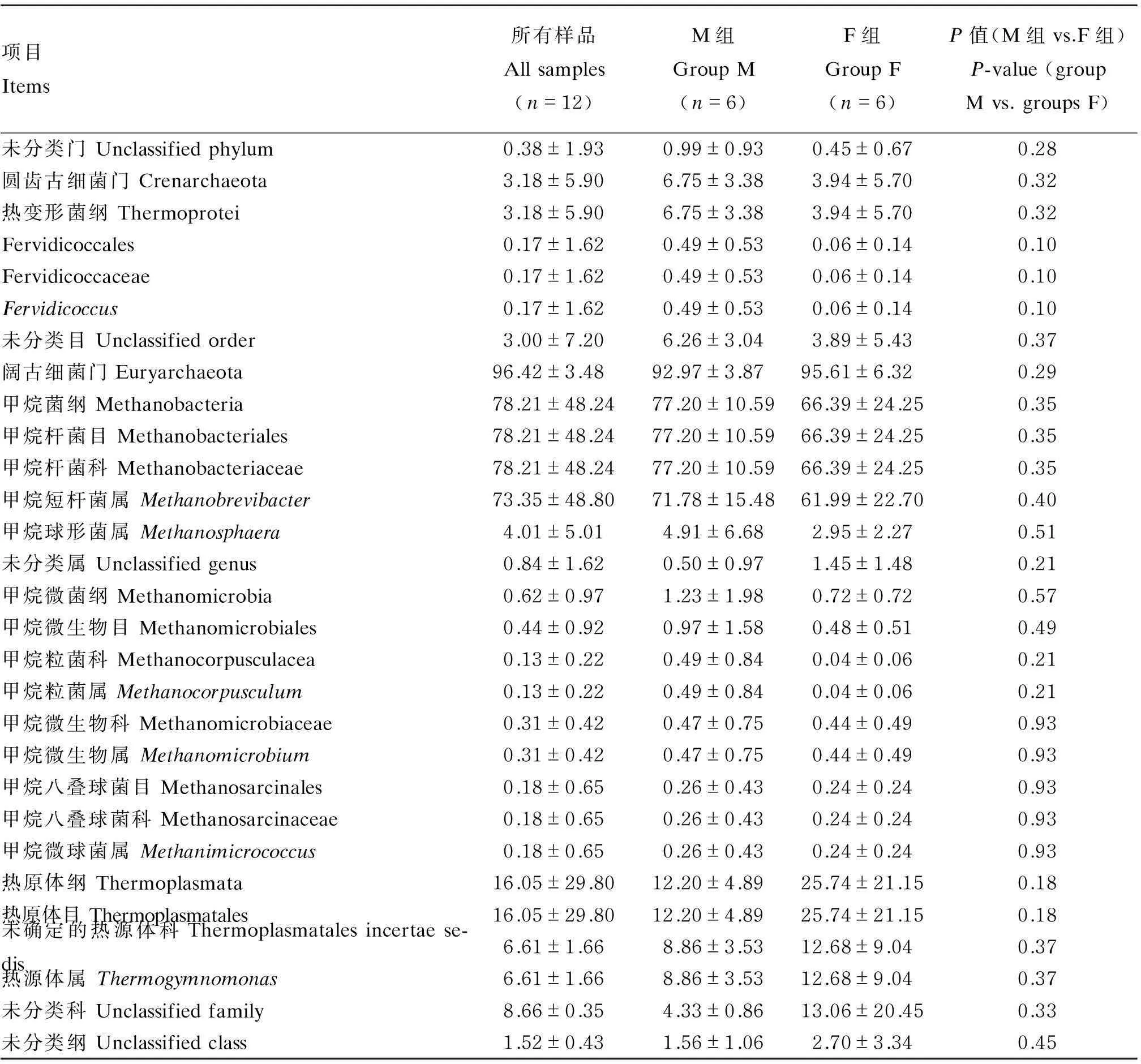
表2 林麝粪便在不同分类水平上的古菌组成及其相对丰度
P<0.05:差异显著 significant difference;P<0.01:差异极显著 extremely significant difference。
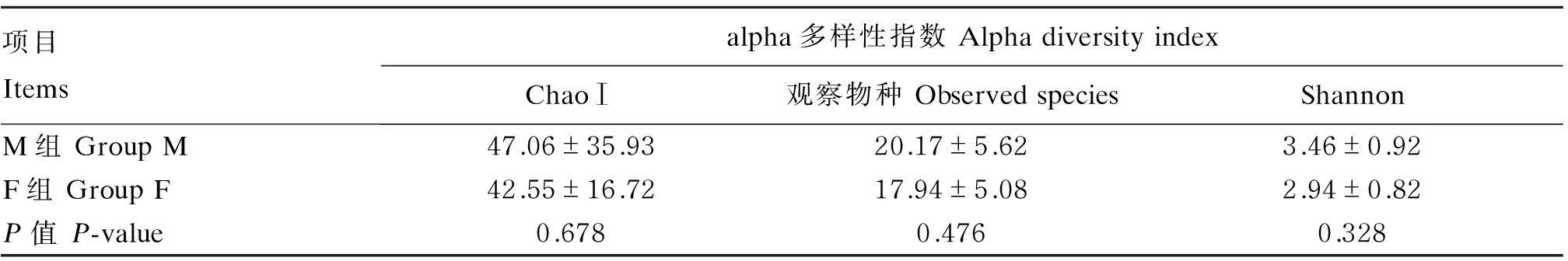
表3 林麝粪便古菌的alpha多样性指数
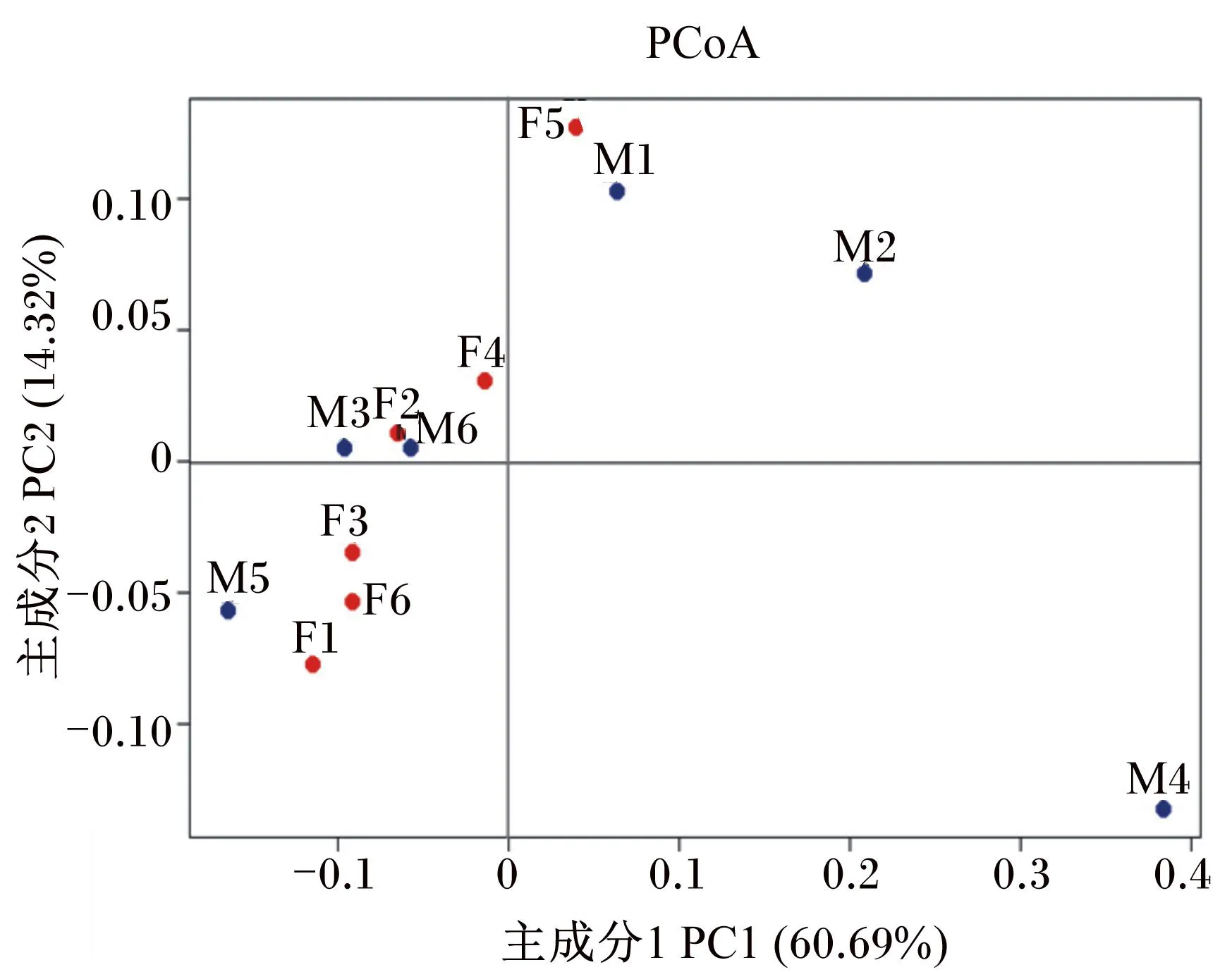
百分数代表了样品受主成分影响下的相似性距离
The percentages represented the similarity distance under influence of PC。
图2林麝粪便古菌PCoA聚类图
Fig.2PCoA diagram reflecting clustering of
fecal archaea of forest musk deer
2.5共享属分析
由于F组和M组在古菌的物种组成和丰度,以及alpha多样性指数上没有显著差异,因此本试验将12个样品作为一个整体,分析了林麝粪便中古菌的共享属。本试验对12个样品间共享的古菌属进行了分析,并根据其丰度以热图的形式进行了展示(图3)。可以看出,12只林麝之间古菌共享属主要来源于Euryarchaeota的Methanobacteria和Thermoplasmata这2个纲。有些共享属在各个样品间的丰度很高,如Methanobrevibacter和Thermogymnomonas,它们的平均相对丰度分别达到了样品古菌总相对丰度的67%和11%。而有些序列虽然也存在于所有样品中,但丰度极低,如Euryarchaeota的未分类纲。各个样品共享属的相对丰度占其古菌总相对丰度的比例平均在96%以上,这说明在属水平上,性别对林麝粪便古菌优势菌群的种类几乎没有影响,但同时应该注意到共享属的丰度在各个样品间存在较大差异。

越偏向红色表示相对丰度越高,越偏向蓝色表示丰度越低。对于不能分类到属水平,但仍然出现在各样品中的共享序列,用其最高分类水平表示。
The more close to red, the higher the relative abundance, while the more close to blue, the lower the relative abundance. Shared sequences were expressed using the highest classification level, which could not be classified to any genus, but still existed in samples.
图3样品间共享属热图
Fig.3Heatmap of shared archaea genera of all samples
3讨论
据了解,本文是第1篇有关林麝胃肠道古菌的研究报道。以往有关动物胃肠道古菌的研究,几乎全部采用的是克隆/测序技术[3-12]。这种技术不仅费时费力,而且仅能鉴定出样品中的优势菌群,不能全面反映动物胃肠道菌群的结构与组成[15]。高通量测序技术,是近年来才采用的一种研究微生物生态学的全新技术,它能较全面地鉴定出样品中微生物的种类[16-17]。在本次研究中,本试验采用高通量测序技术在12头林麝的粪便中,共获得13 130条属于古菌的有效序列,检测到了720个属于古菌的OTU,而以往在反刍动物瘤胃[3-5,7]、袋鼠前肠[6]、动物粪便[8,11]等样品中,一次试验获得的古菌序列数量都在1 000左右,检测到的OTU个数一般都在100个左右,远远小于本次试验结果。本次试验的OTU经行物种注释后,共分为了3个门,8个已知属。以往在动物胃肠道中发现的古菌几乎全部都来源于Euryarchaeota,有关Crenarchaeotea的报道非常罕见,且一次试验最多仅能检测到4~5属的古菌。造成这种差异的原因,应该主要归于试验方法的差异。Kim等[18]综合分析了NCBI等各种公共数据中的古菌序列,并以稀释曲线为基础进行估测,认为瘤胃样品中属于古菌的OTU应该大于1 400个,至少需要24 480条测序序列才能全面覆盖样品中的古菌。本试验的测序序列数和OTU数大概是其1/2,这可能是样品属性不同造成的。因为在前期研究中发现,山羊粪便中古菌多样性显著低于瘤胃[19]。
以往的研究表明,Methanobrevibacter是单胃动物后肠[20-22]和反刍动物瘤胃[5,7]的优势古菌。例如,Wright等[5]在对澳大利亚绵羊瘤胃古菌的研究中就发现,分析所得的65条古菌序列中,有62条都属于Methanobrevibacter。在本试验中,在属水平上,Methanobrevibacter不仅在12个样品间是共享属,而且在雄性、雌性林麝粪便中的相对丰度分别达到了71.78%和61.99%,属于绝对优势核心属,这与以往的很多研究结果是一致的。但也有研究表明,动物胃肠道的优势古菌不是甲烷短杆菌,如Sundset等[23]研究发现,来自驯鹿瘤胃的古菌,53%都属于未鉴定的古菌。Huang等[12]也发现,在生存环境相同且饲喂饲粮一致的条件下,牦牛瘤胃中80.9%的古菌来源于Thermoplasmatales,只有14.9%属于Methanobacteriales。这说明动物胃肠道的古菌在结构和组成上存在宿主特异性,特定动物的古菌结构只有通过实际测定才能确定,而不能根据以往其他动物的试验结果进行推测。
在本试验中,与雄性林麝相比,雌性林麝粪便古菌在从门到属的各级分类水平上,相对丰度都没有显著差异。各项alpha多样性指数,不同性别间也没有显著差异,样品间的遗传距离都较小。这些都说明性别对林麝粪便中古菌的结构和组成没有显著影响。以往研究发现,影响胃肠道古菌的主要因素为品种、饲粮等。例如,Huang等[12]研究发现,与牦牛相比,黄牛瘤胃中Methanobacteriales(21.5% vs. 12.4%)和Methanomicrobiales(9.8% vs. 1.0%)这2个目的相对丰度较高。Zhou等[24]研究发现不同饲料利用效率的肉牛,瘤胃的古菌结构不同,低饲料利用效率组瘤胃古菌多样性要高于高效率组。Min等[25]报道,在肉山羊的瘤胃中,Methanobrevibacter的相对丰度会随饲粮浓缩单宁浓度的增加而线性降低。迄今为止,还未见性别对动物胃肠道古菌影响的相关报道。作者推测,性别本身可能对林麝肠道古菌的结构影响就较小,再加上本次试验中,雄性和雌性饲喂的都是同一饲粮,最终体现出组间没有显著差异。
4结论
在从门到属的各级分类水平上,雄性和雌性林麝粪便中古菌的结构和组成都没有显著差异,Methanobrevibacter是林麝粪便中的优势古菌属。
参考文献:
[1]YANG Q S,MENG X X,XIA L,et al.Conservation status and causes of decline of musk deer (Moschusspp.) in China[J].Biological Conservation,2003,109(3):333-342.
[2]WRIGHT A D G,MA X L,OBISPO N E,et al.Methanobrevibacterphylotypes are the dominant methanogens in sheep from Venezuela[J].Microbial Ecology,2008,56(2):390-394.
[3]CHAUDHARY P P,SIROHI S K,SAXENA J,et al.Diversity analysis of methanogens in rumen ofBubalusbubalisby 16S riboprinting and sequence analysis[J].Gene,2012,493(1):13-17.
[4]SINGH K M,TRIPATHI A K,PANDYA P R,et al.Methanogen diversity in the rumen of Indian Surti buffalo (Bubalusbubalis),assessed by 16S rDNA analysis[J].Research in Veterinary Science,2012,92(3):451-455.
[5]WRIGHT A D G,WILLIAMS A J,WINDER B,et al.Molecular diversity of rumen methanogens from sheep in western Australia[J].Applied and Environmental Microbiology,2004,70(3):1263-1270.
[6]EVANS P N,HINDS L A,SLY L I,et al.Community composition and density of methanogens in the foregut of the Tammar wallaby (Macropuseugenii)[J].Applied and Environmental Microbiology,2009,75(8):2598-2602.
[7]SHIN E C,CHOI B R,LIM W J,et al.Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence[J].Anaerobe,2004,10(6):313-319.
[8]LUO Y H,WRIGHT A D G,LI Y L,et al.Diversity of methanogens in the hindgut of captive white rhinoceroses,Ceratotheriumsimum[J].BMC Microbiology,2013,13:207.
[9]RASTOGI G,RANADE D R,YEOLE T Y,et al.Molecular analyses of methanogen diversity associated with cattle dung[J].World Journal of Microbiology and Biotechnology,2008,24(12):2973-2979.
[10]PEI C X,MAO S Y,CHENG Y F,et al.Diversity,abundance and novel 16S rRNA gene sequences of methanogens in rumen liquid,solid and epithelium fractions of Jinnan cattle[J].Animal,2010,4(1):20-29.
[11]TURNBULL K L,SMITH R P,ST-PIERRE B,et al.Molecular diversity of methanogens in fecal samples from Bactrian camels (Camelusbactrianus) at two zoos[J].Research in Veterinary Science,2012,93(1):246-249.
[12]HUANG X D,TAN H Y,LONG R J,et al.Comparison of methanogen diversity of yak (Bosgrunniens) and cattle (Bostaurus) from the Qinghai-Tibetan Plateau,China[J].BMC Microbiology,2012,12:237.
[13]FRANK K L,ROGERS D R,OLINS H C,et al.Characterizing the distribution and rates of microbial sulfate reduction at middle valley hydrothermal vents[J].The ISME Journal,2013,7(7):1391-1401.
[14]CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods,2010,7(5):335-336.
[15]HIGHLANDER S K.High throughput sequencing methods for microbiome profiling:application to food animal systems[J].Animal Health Research Reviews,2012,13(1):40-53.
[16]SOGIN M L,MORRISON H G,HUBER J A,et al.Microbial diversity in the deep sea and the underexplored “rare biosphere”[J].Proceedings of the National Academy of Sciences of the United States of America,2006, 103(32):12115-12120.
[17]CAPORASO J G,LAUBER C L,WALTERS W A,et al.Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J].Proceedings of the National Academy of Sciences of the United States of America,2011,108(Suppl.1):4516-4522.
[18]KIM M,MORRISON M,YU Z J,et al.Status of the phylogenetic diversity census of ruminal microbiomes[J].FEMS Microbiology Ecology,2011,76(1):49-63.
[19]王继文,王立志,闫天海,等.山羊瘤胃与粪便微生物多样性[J].动物营养学报,2015,27(8):2259-2571.
[20]WEAVER G A,KRAUSE J A,MILLER T L,et al.Incidence of methanogenic bacteria in a sigmoidoscopy population:an association of methanogenic bacteria and diverticulosis[J].Gut,1986,27(6):698-704.
[21]LEADBETTER J R,BREZNAK J A.Physiological ecology ofMethanobrevibactercuticularissp.nov.andMethanobrevibactercurvatussp.nov.,isolated from the hindgut of the termiteReticulitermesflavipes[J].Applied and Environmental Microbiology,1996,62(10):3620-3631.
[22]LEADBETTER J R,CROSBY L D,BREZNAK J A,et al.Methanobrevibacterfiliformissp.nov.,a filamentous methanogen from termite hindguts[J].Archives of Microbiology,1998,169(4):287-292.
[23]SUNDSET M A,EDWARDS J E,CHENG Y F,et al.Rumen microbial diversity in Svalbard reindeer,with particular emphasis on methanogenic archaea[J].FEMS Microbiology Ecology,2009,70(3):553-562.
[24]ZHOU M,HERNANDEZ-SANABRIA E,GUAN L L.Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies[J]. Applied and Environmental Microbiology,2009,75(20):6524-6533.
[25]MIN B R,SOLAIMAN S,SHANGE R,et al.Gastrointestinal bacterial and methanogenic archaea diversity dynamics associated with condensed tannin-containing pine bark diet in goats using 16S rDNA amplicon pyrosequencing[J].International Journal of Microbiology,2014,2014:141909.
Author, WANG Lizhi, associate professor, E-mail: wanglizhi@aliyun.com
(责任编辑王智航)
Fecal Archaeal Diversity of Artificial Breeding Adult Forest Musk Deer Analyzed by High-Throughput Sequencing Technologies
WANG Lizhi1XU Yiying1CAI Yonghua2
(1. Key Laboratory for Animal Disease-Resistance Nutrition of Ministry of Education, Animal Nutrition Institute of Sichuan Agricultural University, Ya’an 625014, China;2. Sichuan Institute of Musk Deer Breeding, Dujiangyan 611845, China)
Abstract:This experiment was conducted to invest the archaeal structure and composition in feces of artificial breeding adult forest musk deer (FMD) using high-throughput sequencing technologies, and to compare the difference between male and female FMD. Twelve healthy FMD with the age of three years were divided into male group (M) and female group (F) according to their gender (6 deer in each group). The fresh feces were collected and DNA was extracted from them. The archaeal universal primers were used to amplify V4 to V5 regions of archaeal 16S rRNA. The amplifications were high-throughput sequenced with the MiSeq 300PE sequencing platform. The QIIME and other softwares were used to analyze the data. The results showed as follows: from phylum to genus levels, the differences of the archaeal relative abundance between group M and group F were not significant (P>0.05). Genetic distance within the samples in group F and group M was 0.16±0.03 and 0.27±0.06, respectively, and that between groups was 0.24±0.07; all samples shared high similarity. The archaea in the feces of FMD can be divided into three phyla, the dominant phylum was Euryarchaeota; on the genus level they can be divided into seven existing genera, the dominant genus was Methanobrevibacter, the next one was Thermogymnomonas. In conclusion, the gender has no significant effect on the archaeal structure and composition in the feces of FMD, and Methanobrevibacter is the dominant genus.[Chinese Journal of Animal Nutrition, 2016, 28(2):477-484]
Key words:forest musk deer; archaea; methanogen; diversity
中图分类号:S852.6
文献标识码:A
文章编号:1006-267X(2016)02-0477-08
作者简介:王立志(1974—),男,湖北襄阳人,副教授,博士,主要从事动物营养学研究。E-mail: wanglizhi@aliyun.com
基金项目:科技部国际合作项目“畜禽低碳养殖关键调控技术合作研究”(2014DFA32860)
收稿日期:2015-08-01
doi:10.3969/j.issn.1006-267x.2016.02.021
猜你喜欢
土壤学报(2022年3期)2022-08-26
大自然探索(2022年5期)2022-07-11
知识就是力量(2022年6期)2022-06-16
北京大学学报(自然科学版)(2020年3期)2020-06-07
科学导报(2018年14期)2018-05-14
求知导刊(2016年30期)2016-12-03
戏剧之家(2016年22期)2016-11-30
散文百家·下旬刊(2016年9期)2016-11-23
人间(2016年27期)2016-11-11
农业与技术(2016年15期)2016-11-09
