
P-糖蛋白抑制剂在PET显像中的应用研究
2016-05-12 08:10何燕苏晋郑晓霞温莉孙树兰
国际放射医学核医学杂志 2016年1期
何燕 苏晋 郑晓霞 温莉 孙树兰
251700,山东省滨州市中心医院放射科
·论著·
P-糖蛋白抑制剂在PET显像中的应用研究
何燕 苏晋 郑晓霞 温莉 孙树兰
251700,山东省滨州市中心医院放射科
目的 探讨11C-GF120918作为PET显像探针对人体P-糖蛋白(P-gp)和乳腺癌耐药蛋白(BCRP)功能的检测及其意义。方法 化学合成11C-GF120918;给小鼠注射11C-GF120918,使用自动伽玛粒子计数管测定不同时间、不同剂量、小鼠各组织器官中11C-GF120918的放射性强度;并使用高效液相色谱监测30 min后小鼠脑部和血液中11C-GF120918代谢情况;P-gp基因敲除组、BCRP基因敲除组、P-gp/BCRP基因敲除组和野生型对照组小鼠分别注射11C-GF120918,进行PET显像,实时监测小鼠大脑中的11C-GF120918放射强度变化。结果11C-GF120918在小鼠各个组织器官中均有广泛分布,相对于正常小鼠差异有统计学意义(χ2=8.14,P<0.05);且11CGF120918代谢稳定,注射后30 min,在大脑和血液中仍有(99.3±0.5)%和(83.2±3.5)%未被代谢,具有较好的生物化学稳定性和辐射稳定性。P-gp/BCRP基因敲除组小鼠大脑中11C-GF120918放射性强度是野生型对照组的9倍(χ2=7.69,P<0.05),而BCRP基因敲除组小鼠大脑中放射性强度是野生型对照组的3倍(χ2=8.24,P<0.05),差异有统计学意义。且11C-GF120918标记效果比较稳定。结论 使用11C-GF120918作为PET显像探针可以用来评价P-gp和BCRP的耐药功能。
P-糖蛋白;肿瘤;正电子发射断层显像术;乳腺癌耐药蛋白
肿瘤细胞多药耐药性(multi-drug resistance,MDR)的产生是肿瘤化疗失败的主要原因,严重降低癌症患者的生存及生活质量。临床上MDR的产生一般由ATP结合盒式蛋白转运蛋白超家族引起,它是一种膜蛋白,通过ATP水解供能将化疗药物排出细胞,引发肿瘤的抗药性[1]。P-糖蛋白(P-gly-coprotein,P-gp)和乳腺癌耐药蛋白(breast cancer resistant protein,BCRP)是ATP结合盒式蛋白转运蛋白超家族成员,这两种蛋白的过量表达参与肿瘤细胞对化疗的耐受[2]。目前,临床上对P-gp和BCRP的检测主要依赖于体外的定性、定量分析,无法在活体体内监测其表达与分布变化,这在一定程度上限制了肿瘤患者化疗前后MDR的动态检测[3]。P-gp抑制剂的PET显像剂目前正在被研究,此类显像剂将与P-gp结合而不被转运,在过表达的脑组织中表现出放射性增强,与放射性同位素标记的P-gp底物的结合恰恰相反,从而可以反映脑组织中P-gp水平[4-5]。本研究利用P-gp抑制剂11C-GF120918监测P-gp和BCRP在小鼠体内的表达与分布,评价其应用价值。
1 材料与方法
1.1 试剂与仪器
GF120918和11C-CH3I购自沈阳宝希迪商贸有限公司。异氟烷、吐温80、二甲基甲酰胺(N,N-dimethylformamide,DMF)和羟基四丁胺购自Aladdin-阿拉丁试剂(上海)有限公司。应用日本岛津LC-20A高效液相色谱仪,制备柱使用日本Capcell Pak公司的C18 UG 80色谱柱(10 mm×250 mm),分析柱使用美国Nova-Pak公司的C18柱(100 mm×8 mm)。其他仪器包括:自动伽玛粒子计数管(Wizard 3″1480,PerkinElmer,美国)、离心机(MX-105,托弥公司,日本)和PET仪(西门子公司,德国)。
正常小鼠55只,P-gp基因敲除鼠、BCRP基因敲除鼠和P-gp/BCRP基因敲除鼠各10只,均购自北京利昊生物科技有限公司,体重20~30 g,年龄2个月。
1.211C-GF120918合成
在0.35 ml溶有1.0 mg GF120918的DMF溶液中加入7 μl 0.33 mol/L羟基四丁胺的甲醇溶液,之后加入11C-CH3I,90℃反应5 min(图1),冷却后加入0.5 ml洗脱液,洗脱液成分为乙腈、水和三乙胺(60∶40∶0.1,V/V/V)。制备方法:洗脱液等度洗脱,洗脱流速4 ml/min,254 nm紫外检测器和辐射检测器检测,GF120918和11C-GF120918的保留时间分别为3.0~5.0 min和9.0~10 min;收集9.0~10 min组分,相同条件再次过柱纯化。生理盐水加吐温80(1 μl/ml)透析纯化产物,即得到可用于动物实验的11C-GF120918。

图1 11C-GF120918的合成 图中,DMF:二甲基甲酰胺。Fig.1 The synthesis of11C-GF120918
1.3 检测11C-GF120918在小鼠体内的分布
正常小鼠20只,通过静脉注射0.25 mg/kg11CGF120918,分别于5、15、30、60 min后断颈法处死小鼠,分析检测其在小鼠体内的组织分布情况。
正常小鼠每组5只,分为7组,分别注射0.25mg/kg11C-GF120918,同时每组分别注射0、0.1、0.5、1.0、3.0、5.0、10.0mg/kgGF120918,以未注射GF120918小鼠为对照组,30 min后断颈法处死,检测其在小鼠体内的组织分布情况。
通过检测小鼠的大脑、血液、心脏、肝脏、脾脏、肾脏和肌肉在5、15、30 min和60 min时的放射性强度,获得11C-GF120918在小鼠体内的分布情况。使用自动伽玛粒子计数管测定放射性强度。放射性强度=[某组织放射性强度(MBq)/某单位组织样品(g)]/[注射剂量(MBq)/小鼠体重(g)]。
1.411C-GF120918在小鼠大脑和血液中的代谢
P-gp基因敲除组、BCRP基因敲除组、P-gp/ BCRP基因敲除组和野生型对照组小鼠,每组5只,分别注射0.015 mg/kg11C-GF120918,30 min后断颈法处死,解剖获得小鼠大脑,心脏取血。血液在4℃下经13 000×g离心3 min,取上层血清立即加入等体积的冰冷乙腈去蛋白,然后经20 000×g离心2 min,取上清。脑组织研碎溶于生理盐水中,加入等体积的乙腈,然后经20 000×g离心2 min,收集上清。
采用高效液相色谱法检测上清液中11CGF120918与11C代谢物含量,具体方法:洗脱液等度洗脱,洗脱液为乙腈与50 mmol/L醋酸钠盐缓冲溶液(pH 4.7,45∶55,V/V)混合液,11C-GF120918与11C代谢物的保留时间分别为6.2 min和2.2 min。检测11C-GF120918与11C代谢物,峰面积积分计算其含量。
1.5 小鼠11C-GF120918的PET显像
P-gp基因敲除组、BCRP基因敲除组、P-gp/ BCRP基因敲除组和野生型对照组小鼠,每组5只。使用异氟烷麻醉小鼠,静脉注射0.25 mg/kg11CGF120918,将小鼠背部朝上置于扫描床上,对小鼠脑部进行PET图像采集,采集时间为11C-GF120918注射后115~120 min。图像采集条件:以二维模式进行PET图像采集,采集完成后使用机器自动对PET图像进行衰减校正。图像重建采用有序子集最大期望值迭代法。用PETDyhamicsystem软件自动勾画出60 min放射性活性——时间动态曲线图,使用0~60 min脑曲线下面积[AUCbrain(0-60)min]表征总放射性强度。
1.6 数据处理
2 结果
2.111C-GF120918在小鼠体内的组织分布
不同时间小鼠不同组织器官的11C-GF120918放射性强度见表1。由表1可知,小鼠注射11CGF120918后,小鼠大脑摄取11C-GF120918剂量最少;小鼠血液中11C-GF120918的水平下降很快,15 min时11C-GF120918水平与5 min时相比,差异有统计学意义(χ2=7.56,P<0.05);心脏和肌肉中的摄取剂量呈降低趋势,30min时11C-GF120918水平与5 min时相比,差异有统计学意义(χ2=7.61和6.47,P<0.05);肝脏中11C-GF120918的摄取呈先升高后降低的趋势;脾脏对11C-GF120918的摄取在注射15 min后才开始缓慢上升,最后维持在一个相对稳定的水平,30 min时与5 min时的放射性强度比较,差异有统计学意义(χ2=7.79,P<0.05)。
表1 不同时间时小鼠各组织器官的11C-GF120918放射性强度(±s)Table 1 The radioactivegy intensitu of11C-GF120918 in mouses at different time and different organs(±s)
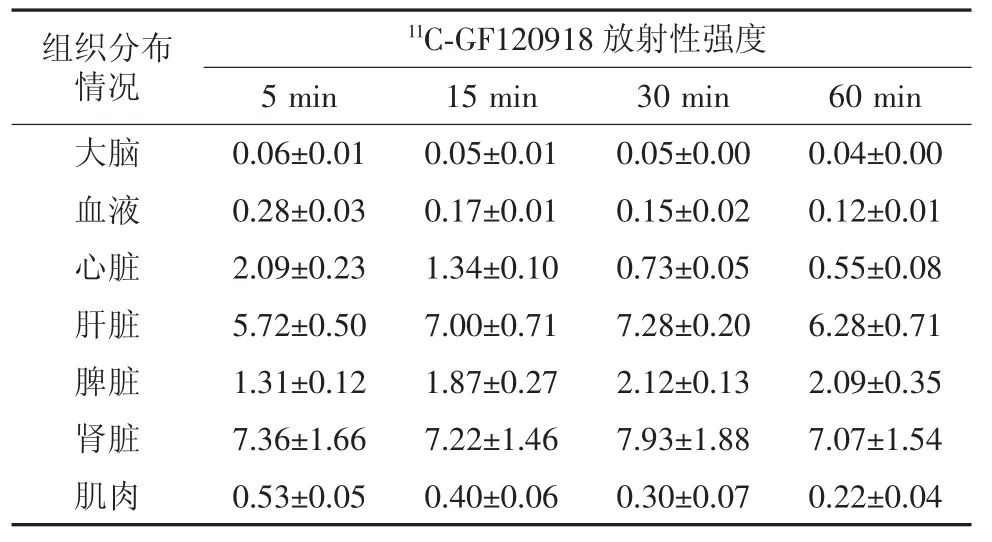
表1 不同时间时小鼠各组织器官的11C-GF120918放射性强度(±s)Table 1 The radioactivegy intensitu of11C-GF120918 in mouses at different time and different organs(±s)
组织分布情况11C-GF120918放射性强度5 min 15 min 30 min 60 min大脑 0.06±0.01 0.05±0.01 0.05±0.00 0.04±0.00血液 0.28±0.03 0.17±0.01 0.15±0.02 0.12±0.01心脏 2.09±0.23 1.34±0.10 0.73±0.05 0.55±0.08肝脏 5.72±0.50 7.00±0.71 7.28±0.20 6.28±0.71脾脏 1.31±0.12 1.87±0.27 2.12±0.13 2.09±0.35肾脏 7.36±1.66 7.22±1.46 7.93±1.88 7.07±1.54肌肉 0.53±0.05 0.40±0.06 0.30±0.07 0.22±0.04
不同GF120918注射剂量下小鼠各组织器官的11C-GF120918放射性强度见表2。由表2可知,小鼠各组织器官对GF120918呈现不同的剂量依赖性,当注射剂量>0.5 mg/kg,大脑对11C-GF120918的摄取显著升高,相对于对照组差异有统计学意义(χ2=8.56,P<0.05);血液中11CGF120918水平不受注射剂量的影响,维持稳定;心脏和肌肉中11C-GF120918的摄取都明显随注射剂量的增加而升高;肝脏和肾脏随注射剂量的增加而降低。
2.2 小鼠大脑和血液中11C-GF120918的代谢情况
野生型对照组小鼠体内注射11C-GF120918后30 min,大脑和血液中分别有(95.4±1.7)%和(95.8± 1.9)%的11C-GF120918未被代谢,而对于 P-gp/ BCRP基因敲除组小鼠大脑和血液中则分别有(99.3±0.5)%和(83.2±3.5)%未被代谢。在血液中,野生型对照组未代谢的11C-GF120918量高于BCRP基因敲除组和P-gp基因敲除组。
表2 不同GF120918注射剂量小鼠各组织器官的11C-GF120918放射性强度(±s)Table 2 The radioactiveity intensity of11C-GF120918 in mouse′s different organs with different GF120918 injected doses(±s)
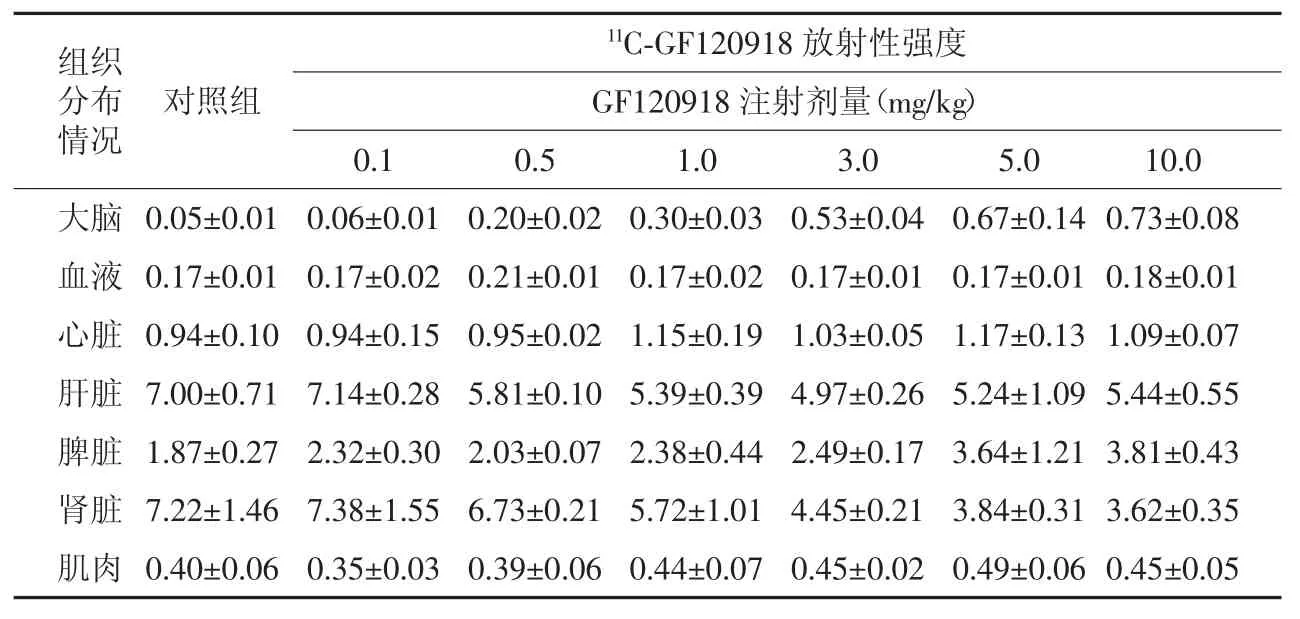
表2 不同GF120918注射剂量小鼠各组织器官的11C-GF120918放射性强度(±s)Table 2 The radioactiveity intensity of11C-GF120918 in mouse′s different organs with different GF120918 injected doses(±s)
组织分布情况11C-GF120918放射性强度GF120918注射剂量(mg/kg)0.1 0.5 1.0 3.0 5.0 10.0大脑 0.05±0.01 0.06±0.01 0.20±0.02 0.30±0.03 0.53±0.04 0.67±0.14 0.73±0.08血液 0.17±0.01 0.17±0.02 0.21±0.01 0.17±0.02 0.17±0.01 0.17±0.01 0.18±0.01心脏 0.94±0.10 0.94±0.15 0.95±0.02 1.15±0.19 1.03±0.05 1.17±0.13 1.09±0.07肝脏 7.00±0.71 7.14±0.28 5.81±0.10 5.39±0.39 4.97±0.26 5.24±1.09 5.44±0.55脾脏 1.87±0.27 2.32±0.30 2.03±0.07 2.38±0.44 2.49±0.17 3.64±1.21 3.81±0.43肾脏 7.22±1.46 7.38±1.55 6.73±0.21 5.72±1.01 4.45±0.21 3.84±0.31 3.62±0.35肌肉 0.40±0.06 0.35±0.03 0.39±0.06 0.44±0.07 0.45±0.02 0.49±0.06 0.45±0.05对照组
2.3 小鼠的11C-GF120918 PET显像
小鼠注射11C-GF120918后行PET显像,结果发现,野生型对照组(图2中A)和BCRP基因敲除组(图2中C)小鼠大脑部的放射性强度相对较低,而P-gp基因敲除组(图2中B)与P-gp/BCRP基因敲除组(图2中D)的小鼠放射性强度相对较高。
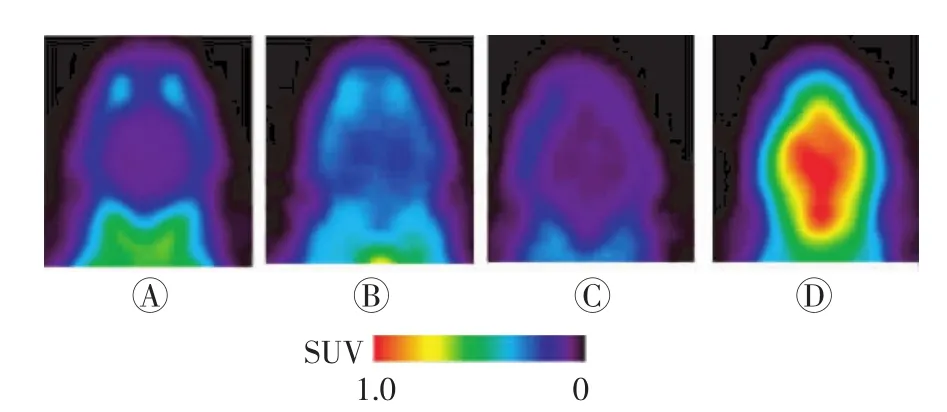
图2 11C-GF120918在小鼠脑部的PET显像 图中,A:野生型对照组;B:P-gp基因敲除组;C:BCRP基因敲除组;D:P-gp/BCRP基因敲除组。Fig.2 The PET imaging of11C-GF120918 in mouse′s brain
同时,注射11C-GF120918后,P-gp/BCRP基因敲除组的摄取剂量先上升后平稳,而P-gp基因敲除组、BCRP基因敲除组和野生型对照组对11CGF120918的摄取是注射后立即下降然后平稳。对比平稳时的数据发现,P-gp/BCRP基因敲除组和P-gp基因敲除组的摄取分别是野生型对照组的9倍和3倍,差异具有统计学意义(χ2=7.69和8.24,P<0.05);BCRP基因敲除组与野生型对照组差异无统计学意义(图3)。

图3 不同时间小鼠大脑11C-GF120918放射性强度变化 图中,P-gp:P-糖蛋白;BCRP:乳腺癌耐药蛋白。Fig.3 The radioactivity intensity of11C-GF120918 in mouse′s brain at different time
对图3曲线进行积分计算总放射性强度,其中P-gp/BCRP基因敲除组60 min内[AUCbrain(0~60min)]总放射性强度为61.4±3.7、P-gp基因敲除组为16.8± 2.9、BCRP基因敲除组为5.9±0.5、野生型对照组为6.5±1.1。P-gp/BCRP基因敲除组与P-gp基因敲除组的总放射性强度分别为野生型对照组的9倍和3倍,差异有统计学意义(χ2=7.82和8.11,P<0.05),BCRP基因敲除组和野生型对照组的总放射性强度无明显差异。
3 讨论
P-gp是ABC转运蛋白超家族成员之一,2009年Aller等[6]就发表了有关P-gp晶体结构的研究文章,目前临床上对P-gp的检测主要还是依赖病理组织在体外的定性、定量分析,无法在活体体内检测P-gp的表达与分布情况,这在一定程度上限制了肿瘤患者化疗前后MDR的动态检测。BCRP又称半转运蛋白,由一个C端的跨膜结构域和一个N端的核苷酸结构域组成。BCRP主要分布于脑、小肠和肝脏等正常组织和肿瘤细胞表面[7],在肿瘤细胞中过表达,帮助肿瘤细胞外排药物,是一种干细胞检测标志物[8]。
本研究利用一种P-gp抑制剂11C-GF120918,研究P-gp和BCRP的药物外排功能,通过正电子核素标记来放大信号,产生类似PET显像探针的作用,并评价11C-GF120918探针在小鼠体内的组织分布以及代谢情况,为未来肿瘤患者MDR的改善以及化疗方法的优化提供重要的临床依据。
本研究在野生型小鼠体内注射11C-GF120918,30 min时大脑的摄取最少,并且很快达到平稳,11C-GF120918可以迅速从大脑中排出,这些结果表明该探针可被大脑的血脑屏障外排,且与11CLaniquidar注射后的情况一致。11C-Laniquidar是一种第三代MDR抑制剂,大脑摄取较低,所以在示踪剂中可以当作一种底物而不是抑制剂[9]。因此,本研究认为,11C-GF120918也可以被当作底物,并且注射后30 min是PET显像的合适时间。
当11C-GF120918注射剂量增加时,血液中的水平不会发生明显变化,因为其他组织会摄取血液中11C-GF120918,并且也会被肝脏、肾脏代谢,使血液中11C-GF120918的水平相对稳定,然而,这又同时导致大脑、心脏等组织中的11C-GF120918水平升高,即便存在血脑屏障的外排,但这种作用也是有限度的,另外,肝脏、肾脏可以代谢11CGF120918,所以注射剂量增加后,肝脏、肾脏中的11C-GF120918也不会明显升高。当11C-GF120918注射剂量>0.5 mg/kg时,其在大脑中的摄取也随之增加,并且P-gp/BCRP基因敲除组小鼠的总放射性强度是野生型对照组的9倍,这些结果表明11CGF120918在大脑中的渗透情况受P-gp和BCRP的调节,因此,体内11C-GF120918的放射性强度可以作为评价P-gp和BCRP功能的指标,并且,当注射剂量>3.0 mg/kg时可以应用于PET显像。
虽然11C-GF120918与BCRP的功能有关,但野生型对照组与BCRP基因敲除组的小鼠大脑中放射性强度不随时间改变,两组的总放射性强度也几乎是相同的。然而P-gp基因敲除组的总放射性强度是野生型对照组的3倍,却是P-gp/BCRP基因敲除组的1/4。这些结果可以解释为:因P-gp基因敲除组的BCRP mRNA水平是野生型对照组的3倍[10],所以P-gp基因敲除组中BCRP会抑制11C-GF120918的渗透,因而P-gp基因敲除组的总放射性强度是P-gp/BCRP基因敲除组的1/4;P-gp和BCRP都是小鼠血脑屏障中重要的外流转运蛋白,从而保护大脑,P-gp基因敲除的小鼠会上调BCRP的表达[11];野生组与BCRP基因敲除组结果相似,或许可以表明在限制11C-GF120918渗透的过程中,P-gp比BCRP起了更重要的作用。近期文献报道,在研究与P-gp和BCRP功能相关的大脑渗透探针时,P-gp/BCRP基因敲除组小鼠比P-gp或BCRP基因敲除组能更有效地放大与显示PET显像效果,从而应用于临床预警疾病[12]。
本研究中,野生型对照组中未代谢11CGF120918的量比P-gp/BCRP基因敲除组多12%,这表明11C-GF120918在肝肾中进行了新陈代谢并不是立即被P-gp或BCRP外排。也正是由于血脑屏障使得只有一小部分疏水的代谢物进入大脑,所以P-gp/BCRP基因敲除组的小鼠才会有一个相对较高的未代谢11C-GF120918量。
本研究创新之处在于使用PET显像探针11CGF120918用于P-gp和BCRP与大脑渗透作用相关功能的研究,结果表明,该探针具有相对较高的生化稳定性和放射性强度,可以较准确地反映P-gp和BCRP的水平和分布情况,并可用于PET体内显像评价P-gp功能,有助于指导肿瘤患者个体化用药,在未来临床研究和相关疾病的预防中有巨大潜力。
利益冲突 本研究由署名作者按以下贡献声明独立开展,不涉及任何利益冲突。
作者贡献声明 何燕负责方法建立、论文撰写及其后期审阅工作;苏晋,郑晓霞 温莉,孙树兰负责现场实验、数据分析等工作。
[1]应帅,郑婷婷,陈培远,等.BCRP/ABCG2的结构功能及相关抑制剂研究[J].中国医药生物技术,2013,8(3):201-205.DOI: 10.3969/cmba.j.issn.1673-713X.2013.03.009. Ying S,Zheng TT,Chen PY,et al.Study on the structure and functionofBCRP/ABCG2anditsrelatedinhibitors[J].ChinMedBiotechnol,2013,8(3):201-205.
[2]Luurtsema G,Schuit RC,Klok RP,et al.Evaluation of[11C]laniquidar as a tracer of P-glycoprotein:radiosynthesis and biodistribution in rats[J].Nucl Med Biol,2009,36(6):643-649.DOI: 10.1016/j.nucmedbio.2009.03.004.
[3]Zhou S,Schuetz JD,Bunting KD,et al.The ABC transporter Bcrp1/ ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype[J].Nat Med, 2001,7(9):1028-1034.DOI:10.1038/nm0901-1028.
[4]Hubensack M,Mueller C,Hoecherl PA,et al.Effect of the ABCB1 modulators elacridar and tariquidar on the distribution of paclitaxel in nude mice[J].J Cancer Res Clin Oncol,2008,134(5):597-607. DOI:10.1007/s00432-007-0323-9.
[5]Ikoma Y,Takano A,Ito H,et al.Quantitative analysis of11C-verapamil transfer at the human blood-brain barrier for evaluation of P-glycoprotein function[J].J Nucl Med,2006,47(9):1531-1537.
[6]Aller SG,Yu J,Ward A,et al.Structure of P-Glycoprotein reveals a molecular basis for Poly-Specific drug binding[J].Science,2009, 323(5922):1718-1722.DOI:10.1126/science.1168750.
[7]Maliepaard M,Scheffer GL,Faneyte IF,et al.Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues[J].Cancer Res,2001,61(8):3458-3464.
[8]郁春景,吴翼伟,万卫星.P-gp功能的PET显像剂的研究进展[J].国际放射医学核医学杂志,2014,38(6):403-407.DOI:10.3760/cma.j.issn.1673-4114.2014.06.013. Yu CJ,Wu YW,Wan WX.Progress in PET imaging evaluating of P-gp function[J].Inter J Radiat Med Nucl Med,2014,38(6):403-407.
[9]戴春玲,吴翼伟,万卫星.P-gp功能的PET显像剂的研究进展[J].中国药理学通报,2005,21(5):513-518.DOI:10.3321/ j.issn:1001-1978.2005.05.001. Dai CL,Wu YW,Wan WX.The development of the reversal of the tumormultidrugfesistance[J].ChinPharmacolBull,2005,21(5):513-518.
[10]Nicolazzo JA,Katneni K.Drug transport across the blood-brain barrier and the impact of breast cancer resistance protein(ABCG2)[J].Curr Top Med Chem,2009,9(2):130-147.DOI:10.2174/156802609787521580.
[11]Jonker JW,Freeman J,Bolscher E,et al.Contribution of the ABC transporters Bcrp1 and Mdr1a/1b to the side population phenotype in mammary gland and bone marrow of mice[J].Stem Cells,2005,23(8):1059-1065.DOI:10.1634/stemcells.2005-0150.
[12]Kawamura K,Yamasaki T,Yui J,et al.In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using[11C]gefitinib[J].Nucl Med Biol,2009,36(3):239-246.DOI:10.1016/j.nucmedbio.2008.12.006.
Developing P-glycoprote ininhibitor markedby PET
HeYan,SuJin,ZhengXiaoxia,WenLi,SunShulan
Department of Radiology,Binzhou City Center Hospital of Shandong Province,Binzhou 251700,China
He Yan,Email:495796192@qq.com
Objective To explore a PET probe,11C-GF120918 in the assessing of the function and significance of P-glycoprotein(P-gp)and breast cancer resistance protein(BCRP).MethodsThe mice were injected with chemically synthesized11C-GF120918.An automatic gamma counter was used to measure the11C-GF120918 radiation intensity of the various organs of the mice at different times and dosages.Simultaneously,HPLC was employed to detect the metabolism of11C-GF120918 in the brain and blood of the mice.The four mice groups,namely,P-gp knockdown mice,BCRP knockdown mice,P-gp/ BCRP knockdown mice,and wild mice,were manually injected with11C-GF120918.The radiation intensity of11C-GF120918 in the mice brain was detected by PET.ResultsAfter the11C-GF120918 injection,the tissues and organs of mice were more widely distributed compared with those of the wild mice(χ2=8.14,P<0.05).Thirty minutes after injection,the11C-GF120918 radiation intensity in the brain and blood were still(99.3±0.5)%and(83.2±3.5)%,respectively,with better biochemistry and radiation stability.In PET studies,AUCbrain[0~60min]in the P-gp knockout mice was nine times higher than that in the wild group(χ2=7.69, P<0.05).The AUCbrain[0-60min]of the BCPR knockout mice was three times higher than that in the wild group(χ2=8.24,P<0.05).The evident effect of11C-GF120918 was relatively stable.Conclusion11CGF120918 can be used as PET probes to evaluate the multi-drug resistance of P-gp and BCRP.
P-glycoprotein;Neoplasms;Positron-emission tomography;Breast cancer resistance protein
何燕,Email:495796192@qq.com
10.3760/cma.j.issn.1673-4114.2016.01.001
2015-04-05)
猜你喜欢
中老年保健(2022年4期)2022-08-22
农村青少年科学探究(2022年4期)2022-07-29
现代临床医学(2021年3期)2021-07-16
中成药(2018年2期)2018-05-09
中成药(2016年8期)2016-05-17
太空探索(2015年10期)2015-07-18
中国药业(2014年12期)2014-06-06
