
回族Lynch综合征一家系错配修复基因表达及突变情况分析
2016-05-10 11:24解澎任景丽汪文杰虎建恩胡桂明吴会芳牛海鸥
山东医药 2016年28期
解澎,任景丽,汪文杰,虎建恩,胡桂明,吴会芳,牛海鸥
(郑州大学第二附属医院,郑州450014)
回族Lynch综合征一家系错配修复基因表达及突变情况分析
解澎,任景丽,汪文杰,虎建恩,胡桂明,吴会芳,牛海鸥
(郑州大学第二附属医院,郑州450014)
目的 分析回族Lynch综合征一家系错配修复基因(MMR)表达及突变情况,探讨回族Lynch综合征的基因特点。方法 先证者男性,37岁,第4代,回族,直肠低分化腺癌。本家系现存6代(均为回族),共有结直肠癌患者14例,在世7例。提取本家系中6例患者及5例正常人外周血及肿瘤组织DNA。采用DNA测序检测肿瘤细胞DNA微卫星不稳定性(MSI),采用免疫组化法检测肿瘤细胞MMR表达,采用PCR技术检测MMR基因突变情况。结果 该家系结直肠癌患者肿瘤组织MSI检测均表现为高度微卫星不稳定性(MSI-H);肿瘤组织MMR家族中MLH1表达均为阴性;MLH1基因第一外显子存在2个新的错义突变位点c.264G>T、c.265G>T,其中c.265G>T突变导致MLH1基因蛋白质翻译在该位点提前终止。结论 回族Lynch综合征家系存在MLH1基因突变,即存在第一外显子错义突变位点c.265G>T。
Lynch综合征;结直肠肿瘤;错配修复基因;微卫星不稳定性;回族
Lynch综合征为常染色体显性遗传疾病,发病率占所有结直肠癌的2%~3%[1],其主要发病机制为错配修复基因(MMR)突变导致的DNA错配修复功能障碍。Lynch等[1]最早确定MMR基因为癌症的易感基因,随后越来越多的错配修复家族基因被发现,目前报道的主要有MLH1、MSH2、MSH6、PMS1、PMS2等[2]。我国对Lynch综合征的研究起步较晚,针对少数民族家系的研究更少。本课题组在临床工作中发现1个回族Lynch综合征家系,现对该家系的基因情况进行分析,探讨其基因特点。
1 资料与方法
1.1 临床资料 先证者(第4代,Ⅳ8),男性,37岁,回族,肠镜检查见距肛门8~13 cm直肠左前壁有一菜花样肿物。术后病理诊断为直肠低分化腺癌。本家系现存6代(均为回族),共有结直肠癌患者14例,其中结肠癌12例,直肠癌3例(1例为直肠癌术后患结肠癌)。至2016年1月尚在世7例;结肠癌发病部位均为右半结肠,病理诊断为管状腺癌或黏液腺癌,组织分化程度均较低,无原位癌。均行右端结肠切除术,术后均行常规化疗。家系中存在3例患者互为1级亲属的现象。家族中连续3代均有发病。该家系符合Amsterdam标准。其中1例因年龄大、身体状况极差未纳入本研究,余6例患者的家系位置及临床资料见表1。
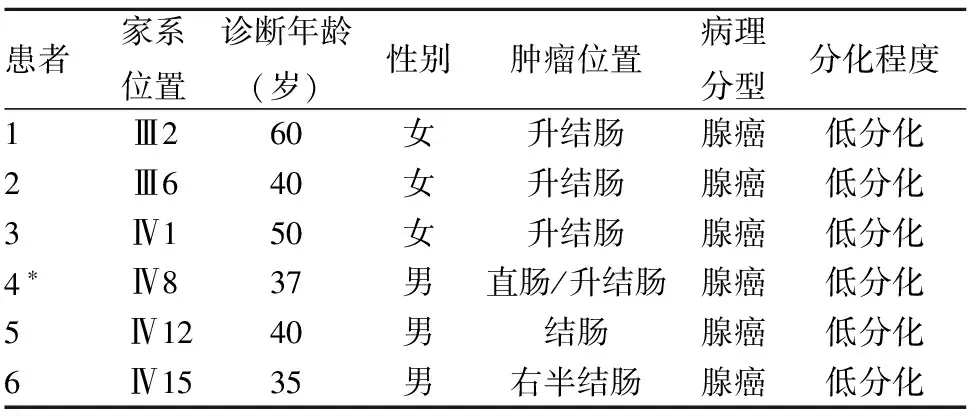
表1 6例结直肠癌患者的基本资料
注:*为先证者;Ⅲ为第三代,Ⅳ为第四代。
1.2 外周血及肿瘤组织DNA提取 取本家系中6例患者及5例正常人的外周血,采用QIAGEN公司全血DNA提取试剂盒提取外周血DNA,置于-20 ℃冰箱保存。随机选取其中3例患者(Ⅳ1、Ⅳ8、Ⅳ15)既往手术所取的肿瘤石蜡包埋组织,采用QIAGEN公司石蜡包埋组织DNA提取试剂盒提取DNA,置于-20 ℃冰箱保存。
1.3 肿瘤组织微卫星不稳定性(MSI)检测 选取1.2所提DNA作为检测样本,采用直接测序法检测DNA微卫星标记序列是否与UCSC数据库人类基因组序列一致。依照美国国立癌症研究所推荐的判定标准选取5个检测位点,分别为BAT25、BAT26、D5S346、D2S123、D17S150。判定标准:≥2个位点不稳定判定为高度微卫星不稳定(MSI-H),1个位点不稳定判定为低度微卫星不稳定(MSI-L),5个位点均稳定判定为微卫星稳定(MSS)。测序分析结果显示,3例患者肿瘤组织DNA均表现为MSI-H。
1.4 肿瘤组织MMR表达检测 取1.2中3例患者石蜡包埋肿瘤组织,并以远离肿瘤的切缘结肠组织(距肿瘤5 cm以上)作为对照,采用免疫组化法检测肿瘤组织中MLH1、MSH2及MSH6表达。采用半定量评估法判定结果。对染色深度和阳性细胞百分率分别计分:无染色为0分,弱染色为1分,中等强染色为2分,强染色为3分;阳性细胞比例<1%为0分,1%~10%为1分,>10%~50%为2分,>50%~80%为3分,>80%为4分。染色强度和阳性细胞率之和为0~2分为阴性,3~5分为阳性,6~7分为强阳性。结果显示,3例患者肿瘤组织MLH1表达均阴性,MSH2及MSH6表达均阳性。
1.5 MMR突变检测 1.3结果显示3例患者肿瘤组织中MLH1基因表达均缺陷,故对MLH1基因19个外显子进行测序。选取家系先证者全血DNA为样本,应用Primer Premier5.0软件设计引物,引物由上海生工生物工程有限公司合成。采用PCR反应试剂盒对MLH1基因外显子区域进行PCR扩增,反应产物经琼脂糖凝胶电泳并测序。测序结果对比采用SeqMan软件。
该家系先证者外周血DNA在MLH1基因1号外显子上发现有2个突变位点,分别为c.264G>T、c.265G>T,其余外显子未发现突变。在NCBI SNP数据库中未查到上述位点已知的SNP。根据突变序列翻译出氨基酸序列在NCBI Protein Blast数据库中与常见的几种动物氨基酸进行特异性对比,发现这些动物在该位点氨基酸序列与野生型一致,推测该位点所在序列可能为保守序列,即c.264G>T突变为同义突变,不对氨基酸序列产生影响,而c.265G>T可导致MLH1基因序列翻译在第22位提前终止。采用Polyphen进行蛋白功能预测,预测结果显示此突变为有害突变。推测c.265G>T突变由于提前终止氨基酸序列翻译从而使蛋白质功能丧失。
1.6 突变位点家系内验证 参照1.5方法再次检测该家系内5例患者及另外5例正常个体外周血MLH1基因1号外显子突变情况,反应体系及条件均同1.5。结果显示,5例患者均存在c.265G>T突变,正常个体中4例未发现突变、1例发现相同突变。见图1。
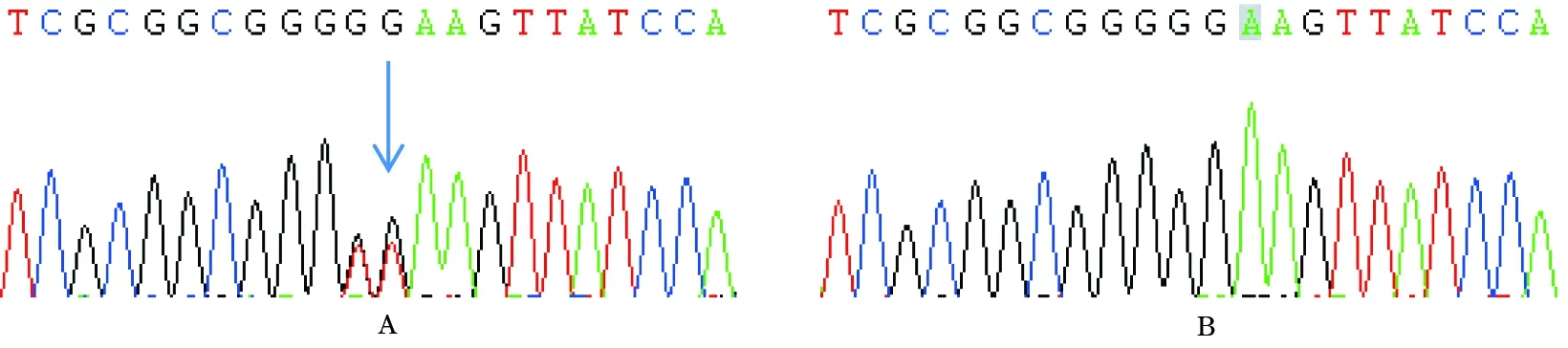
注:A为结直肠癌患者,B为正常人;箭头所指为MLH1基因1号外显子c.265G>T突变。
图1 家系内结直肠癌患者及正常人MLH1基因1号外显子测序图
2 讨论
目前对Lynch综合征主要依靠基因检测结合临床诊断[3,4]。2015年美国临床肿瘤学会(ASCO)指出,疑似Lynch综合征患者均应进行MMR蛋白免疫组化检测或MSI分析,若MMR蛋白缺失或MSI-H,则应排除BRAF V600E突变和MLH1启动子甲基化因素,而后进行MMR基因种系突变检测[5]。
微卫星是指基因组内短序列的重复单元,在DNA复制过程中易发生错误[6]。MSI检测是目前Lynch综合征的初筛手段之一[7],对Lynch综合征患者选择化疗方案和判断预后亦有一定的指导意义[8]。研究表明,处于Ⅱ期MSI-H的Lynch综合征患者不能从5-氟尿嘧啶辅助化疗中获益,该化疗方案会降低患者的生活质量[9]。
MLH1基因是Lynch综合征中突变率最高的MMR基因[10],定位于3p21.3~23,含有19个外显子,编码蛋白含756个氨基酸,通常与其他MMR基因组成复合体发挥作用[11]。李晓芬等[12]统计分析了国内近年来报道的Lynch家系基因突变情况,在检出MMR种系突变的141个Lynch综合征家系中,MLH1种系突变75个(53.2%)。不同的MMR基因突变所表现的发病特点并不完全相同。Vasen等[13]研究表明,MSH2突变基因携带者罹患子宫内膜癌的风险(61%)比MLH1突变基因携带者(42%)高。
我国是多民族国家,但针对少数民族Lynch综合征的研究较少。珠珠等[11]研究了云南地区7个Lynch综合征家系,7个家系均存在MMR基因缺失,其中3个汉族家系检出已知致病突变,但在4个少数民族家系中未检测出包括大片段缺失及基因重排在内的致病突变;提示结直肠癌患者的MMR基因表达以及MMR基因突变可能存在民族差异。
本研究报道的Lynch综合征家系包括6代,均为回族,先证者以结肠癌入院,既往有直肠癌病史,采集信息发现高度聚集的癌症家族史,为明确诊断进行MMR蛋白检测,发现肿瘤组织MLH1表达缺失,继而对家系内患者及部分未患病者进行MMR蛋白及基因检测,发现致病突变,即MLH1基因第一外显子错义突变位点c.265G>T。家系内1例未患病成员基因序列存在与患者相同突变,该成员年龄较小(17岁),推测为致病基因突变携带者,其患肿瘤风险较高,应密切监测,定期体检。本家系患者肿瘤组织DNA表现为MSI-H,为错配修复功能缺陷导致基因在复制过程中的错误得不到有效修复,继而在微卫星位点上出现不同于野生型的表型。
总之,回族Lynch综合征家系存在MLH1基因突变,即存在第一外显子错义突变位点c.265G>T;该发现为我国少数民族Lynch综合征基因突变研究增加了新的数据支持。
[1] Lynch HT, Lynch PM, Lanspa SJ, et al. Review of the Lynch syndrome: history, molecular genetics,screening, differential diagnosis, and medicolegal ramifications[J]. Clin Genet, 2009,76(1):1-18.
[2] Cicek MS, Lindor NM, Gallinger S, et al. Quality assessment and correlation of microsatellite instability and immunohistochemical marker among population and clinic-based colorectal tumors result from the colon cancer family registry[J]. J Mol Diagn, 2011,13(3):271-281.
[3] Yuan L, Chi Y, Chen W, et al. Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency[J]. Int J Clin Exp Med, 2015,8(11):20988-21000.
[4] Wolf AL, Buchanan AH, Farkas LM. Historical review of Lynch syndrome[J]. Coloproctol, 2013,33(2):95-110.
[5] Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndrome: american society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology clinical practice guidelines[J]. Clin Oncol, 2015,33(2):209-217.
[6] Sinicrope FA, Sargent DJ. Molecular pathways: microsatellite instability in colorectal cancer: prognostic, predictive,and therapeutic implications[J]. Clin Cancer Res, 2012,18(6):1506-1512.
[7] Karlitz JJ, Sherrill MR, DiGiacomo DV, et al.Factors associated with the performance of extended colonic resection vs segmental resection in early-onset colorectal cancer: a population-based study[J]. Clin Transl Gastroenterol, 2016(7):e163.
[8] Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a perdictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer[J]. N Engl J Med, 2003,349(3):247-257
[9] Steinke V, Engel C, Buttner R, et al. Hereditary nonpolyposiscancer (HNPCC)/Lynch syndrome[J]. Dtsch Arztebl Int, 2013,110(3):32-38.
[10] Zumstein V, Vinzens F, Zettl A, et al. Systematic immunohistochemical screening for Lynch syndrome in colorectal cancer: a single centre experience of 486 patients[J]. Swiss Med Wkly, 2016(146):w14315.
[11] Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer[J]. Hum Mol Genet, 2001,10(7):735-740.
[12] 李晓芬,袁英.中国Lynch综合征的过去、现在和将来[J].中华结直肠疾病电子杂志,2015,4(3):244-249.
[13] Vasen HF, Wignen JT, Menko FH, et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis[J]. Gastroenterology, 1996,110(4):1020-1027.
[14] 珠珠,黄鉴,潘国庆,等.云南省7个Lynch综合征家系中致癌基因突变研究[J].肿瘤防治研究,2015,42(4):389-393.
Pedigree analysis and gene mutation inspection of Lynch syndrome in Hui nationality
XIEPeng,RENJingli,WANGWenjie,HUJian′en,HUGuiming,WUHuifang,NIUHaiou
(1TheSecondAffiliatedHospitalofZhengzhouUniversity,Zhengzhou450014,China)
Objective To investigate the expression and mutations of DNA mismatch repair (MMR) of Lynch syndrome in the members of a Chinese Hui family. Methods The propositus was male, 37 years old, the forth generation, Hui nationality, had rectal poorly differentiated adenocarcinoma. There were six generations existing (Hui nationality), and a total of 14 patients with colorectal cancer were found including 7 living cases. The peripheral blood and tumor tissue DNA was collected from 6 cases of patients and 5 healthy controls. The microsatellite instability (MSI) was detected through DNA sequencing, the expression of MMR protein was measured using immunohistochemistry, and MMR gene mutations was detected by PCR amplification. Results The MSI detection of tumor tissues in the family members demonstrated MSI-H. The immunohistochemistry showed that MLH1 protein was negative. Two new missense mutation sites (c.264G>T and c.265G>T) in the first exon of MLH1 gene were found in 6 patients and 1 normal individual, and c.264G>T mutation led to early termination of MLH1 protein translation on this site. Conclusion There is MLH1 mutation in Hui family with Lynch syndrome, that is the missense muntation site c.264G>T in the first exon of MLH1 gene.
Lynch syndrome; colorectal neoplasms; DNA mismatch repair; microsatellite instability; Hui nationality
河南省医学科技攻关计划项目(201503099)。
解澎(1988-),男,硕士在读,研究方向为消化系统肿瘤发病机制。E-mail: 18239980973@163.com
任景丽(1964-)女,副主任医师,副教授,研究方向为消化系统肿瘤发病机制。E-mail: jingliren123002@126.com
10.3969/j.issn.1002-266X.2016.28.005
R735.3
A
1002-266X(2016)28-0016-03
2016-01-25)
猜你喜欢
特产研究(2022年6期)2023-01-17
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
四川动物(2017年6期)2017-12-12
四川动物(2017年4期)2017-07-31
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
郑州大学学报(医学版)(2015年2期)2015-02-27
