
基于宏基因组学的鸭源样本中坦布苏病毒检测方法的建立
2016-05-10 11:31邓明俊郑小龙
动物医学进展 2016年4期
孙 涛,王 超,邓明俊,郑小龙,王 群,徐 彪*
(1.山东出入境检验检疫局,山东青岛 266002;2.泰安出入境检验检疫局,山东泰安 250014)
基于宏基因组学的鸭源样本中坦布苏病毒检测方法的建立
孙涛1,王超2,邓明俊1,郑小龙1,王群1,徐彪1*
(1.山东出入境检验检疫局,山东青岛 266002;2.泰安出入境检验检疫局,山东泰安 250014)
摘要:利用二代高通量测序技术建立检测鸭源样品中坦布苏病毒的方法。首先将样品过滤和经核酸酶处理,再利用等温链位移扩增(SPIA)技术快速制备样品总cDNA,最后经磁珠纯化和文库构建后,通过Ion Torrent进行高通量测序获得基因组信息。结果显示,通过SPIA扩增和核酸文库制备的优化,可从微量病原样本中获得327 pmol/μL的核酸文库样本。Ion Torrent测序共获得1 725 436条reads,约6.2 M数据,平均109 bp。数据拼接并Blast发现,病原样品的核酸序列可注释鸭坦布苏病毒全部基因组数据,与首次发生在我国的鸭坦布苏病毒BYD-1株参考序列的同源性为100%。将所测病毒序列与其他5种黄病毒科成员进行同源性比对,发现与近3年发生在我国的鸭坦布苏病毒同源性最高,在98%以上。且NS基因进化树分析与鸭坦布苏病毒处于同一分支上,符合鸭坦布苏病毒特征。本研究建立的基于未知病原SPIA扩增结合高通量测序检测方法,可同步获知坦布苏病毒基因的核酸信息。
关键词:宏基因组学;鸭坦布苏病毒;二代测序
新发病毒性流行病已经日益成为畜牧养殖行业的公共问题。受制于常规检测技术的局限性,传统的、基于PCR的分子生物学鉴定方法必须基于已知病毒的基因序列,病原信息常常得不到快速揭示,从而造成了疫情的蔓延和经济损失。及早发现、鉴别未知或新发病原是疾病防控的关键,同时对疾病的临床治疗也具有重要的指导意义。2010年,曹贞贞等[1]最先发现一种导致蛋鸭产蛋大幅下降,后期伴有神经症状的新型病毒病。因该病剖检症状主要呈现卵泡变性、变形,卵泡膜充血、出血,因此首先命名为鸭出血性卵巢炎(Duck hemorrhagic ovaritis,DHO)。后期随着该病的基因组信息被逐渐揭示[2-9],发现其病原和坦布苏病毒亲缘关系最近,才正式将该病病原名称统一为“鸭坦布苏病毒”(Duck Tembusu virus,DTMUV)。
病毒宏基因组学是研究特定环境及复杂样品的技术方法[10],针对鸭坦布苏病毒这类未知或新发病原,可消除传统分子生物学鉴定的局限,快速获得未知样品的基因组信息。本研究即利用病毒宏基因组学理念结合二代高通量测序技术进行未知鸭源样品中坦布苏病毒的检测。
1材料与方法
1.1材料
1.1.1送检样品病毒感染的细胞培养样品为青岛某公司送检,病毒分离未能确定病原,用常规PCR诊断方法亦未能确定。鸭胚成纤维细胞、BKH细胞,山东出入境检验检疫局动物检疫实验室制备保存。
1.1.2分子生物学试剂DNase Ⅰ、RNase A,天根生物技术有限公司产品;Agencourt○RAMPure○RXP Reagent,Thermo公司产品;Dneasy Kit,Rneasy Mini Kit,QIAGen公司产品;Ovation RNA-Seq System V2基因组扩增盒,Nugen公司产品;Ion ShearTMPlus Reagent Kit、Ion X pressTM Barcode X 、Platimun○RPCR SuperMix、Library Amplification Primer Mix、Ion LibraryTaqMan○RqPCR Mix试剂盒,Life Technologies公司产品。
1.1.3主要仪器Step One Plus荧光PCR仪,购自ABI公司;Ion Chef System 核酸前处理仪,Ion Torrent 测序仪,购自美国Thermo公司。
1.2方法
1.2.1未知样品的处理将细胞培养样品冰浴融化,组织样品研磨后离心,12 000 r/min离心5 min,取上清用0.22 μm滤膜过滤,此过程在超净工作台中完成。然后取滤液加入DNase Ⅰ、RNase A各1.5 μL、10×DNase Ⅰbuffer 40 μL,于37℃水浴3 h,以降解样品中宿主游离核酸,然后置75℃水浴10 min,灭活DNase Ⅰ。
1.2.2DNA和RNA的提取处理后的样品均分为2份,200 μL样品用QIAGen DNA提取试剂盒提取DNA,另外200 μL按Rneasy Mini Kit提取试剂盒操作步骤提取RNA。
1.2.3RNA样品的反转录等温链位移扩增每份病毒RNA样品溶解于5 μL的RNase水,参照Nugen公司Ovation RNA-Seq System V2基因组扩增试剂盒的使用说明。第1步:进行第1链合成,向反应体系中加入第1链合成酶0.5 μL,第1链合成缓冲液2.5 μL,65℃ 2 min,进行引物退火;随后进行第1链合成,反应条件为:4℃ 1 min;25℃ 10 min,42℃ 10 min, 70℃ 15 min,4℃保温。第2步:利用Agencourt RNA Clean XP purification 磁珠纯化后,进行第2链合成,反应体系中加入第2链合成酶0.3 μL ,第2链合成缓冲液9.7 μL,反应条件为:4℃ 1 min;25℃ 10 min,50℃ 30 min,80℃ 20 min,4℃保温。第3步:进行等温链位移扩增。向反应体系中加入等温链位移扩增引物10 μL,等温链位移扩增缓冲液20 μL,SPIA扩增酶10 μL,反应条件为: 4℃ 1 min,47℃ 60 min,80℃ 20 min, 4℃结束反应。
1.2.4核酸文库的制备上述样本全部扩增完毕后,用PCR纯化试剂盒进行纯化。分别取100 ng 纯化产物,用Ion ShearTMPlus Reagent Kit进行酶切打断5 min,酶切产物加入1.8倍体积AMPure○RXP Beads纯化,然后分别加入不同的 Ion X pressTMBarcode接头,E-Gel胶选择片段(约400 bp)构建文库。用Platimun○RPCR扩增试剂盒进行文库扩增,Ion LibraryTaqMan○RqPCR Mix鉴定文库质量。
1.2.5Ion Torrent 测序取20 μL的工作文库上机Ion Chef System进行油包水PCR和碱裂解法制备单链模板,最后将带有模板的微粒溶液自动点样至314芯片中,上机Ion torrent PGM进行高通量测序。
1.2.6生物信息学分析上机测序后经PGM测序仪自动运行得到的测序结果,解析为Bam的原始文件通过pathogen analyze软件进行拼接分析。同时选取GenBank中下载的坦布苏病毒全基因序列作为参考序列,将测序数据通过Blast比对到参考序列中,通过覆盖参考序列的序列数量、比例以及参考序列被覆盖的长度判断样品中是否有对应的病毒。
1.2.7系统进化分析与遗传距离计算将测得序列(暂定为QD株),通过DNA Star软件的MegAlign模块,对病毒全基因组与GenBank中已发表的坦布苏病毒以及其他黄病毒科成员进行同源性比对,并针对注释的病毒NS5基因构建系统进化树,计算遗传距离。从GenBank下载参考序列的毒株(表1),从GenBank下载NS5基因参考序列(表2)。

表1 从GenBank下载的参考序列毒株及其缩写

表2 从GenBank下载的NS5基因参考序列毒株及其缩写
2结果
2.1核酸文库的鉴定
分别取5 μL 100倍稀释后的扩增样本,使用Ion LibraryTaqMan○RqPCR Mix荧光定量法进行浓度测定,标准品梯度稀释成6.8 pmol/L 0.068 pmol/L的标准品,每个梯度做2个平行,进行实时荧光RT-PCR检测,根据质粒拷贝数与Ct值的相关性,由SDS软件2.1得标准曲线为y=-3.305x+17.322,R2=0.995(图1)。目标检测文库检测浓度约为327 pmol/L。
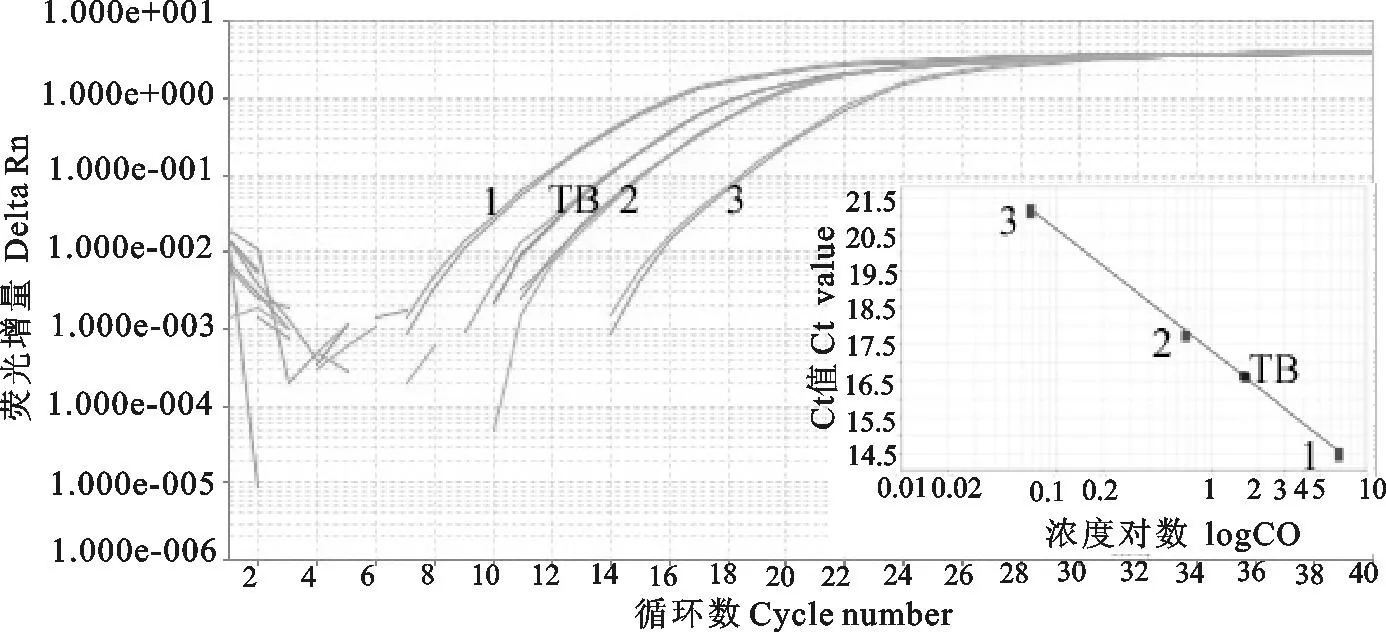
1.6.8 pmol/L标准品扩增曲线;2.0.68 pmol/L标准品扩增曲线 ;3.0.068 pmol/L标准品扩增曲线; TB.目标检测文库扩增曲线
1.Amplification curve of 6.8 pmol/L standard ;2.Amplification curve of 0.68 pmol/L standard;3.Amplification curve of 0.068 pmol/L standard;TB.Amplification curve of target liabrary
图1目标文库及不同稀释度标准样品的扩增曲线
Fig.1Amplification plots of target library and different dilutions of reference standards
2.2Ion Torrent测序
测得6.2 M数据,约3 163 352 reads,匹配reads数为1 725 436(图2)。所测数据经软件自动拼接,同时在NCBI上进行Blast搜索,显示有48段序列涵盖坦布苏病毒全部全基因组序列,与首次发生在我国的鸭坦布苏病毒BYD-1株参考序列[4]的同源性为100%(GenBank 登录号为JF312912)(图3)。
2.3生物信息学分析
将本试验测得的坦布苏病毒QD株全基因序列与其他22株黄病毒科病毒进行同源性比对,发现该株病毒与黄病毒科中的黄热病毒(Yellow fever virus)、登革热病毒(Dengue virus)、伊利乌斯脑炎病毒(Ilheus virus)、西尼罗病毒(West Nile virus)、巴格扎病毒(Bagaza virus)、坦布苏病毒(Tembusu virus)具有亲缘关系,其中与近3年发生在我国的鸭坦布苏病毒同源性最高,达98%以上,与黄热病毒亲缘关系最远(图4)。
国际上,通常以黄病毒NS5基因3′端长约1 kb的区域同源性作为黄病毒属的病毒分类依据[11]。据此,我们将本试验测得的NS5基因与其他黄病毒病毒采用DNA Star进行核苷酸同源性比对,发现与坦布苏病毒处于同一分支上(图5)。

图2 Ion Torrent 测序仪读取序列数

图3 序列搜索比对结果

图4 QD株与其他GenBank已公布序列的核苷酸同源性比较
3讨论
所谓病毒宏基因组学就是通过各种手段将某一复杂样本中所有病毒同其他宿主及微生物区分开来,然后将病毒组的核酸文库同现有的数据库进行比对,从而获得样本中整体病毒组的构成[10]。这种研究手段避开了传统的依据已知核酸序列的研究方法,而直接获取病毒群落的信息,从而获得了复杂样本中更为丰富的病毒组构成,为预防新疾病出现和已知病毒的变异提供了重要的警示作用。Ge X等[12]与杨凡力等[13]就曾在研究中国蝙蝠带毒的自然本底时,应用宏基因组学分析技术注释到了36个病毒科,建立了野生动物源人兽共患病的监测方法。韩文等[14]利用宏基因组学理念,通过大规模测序及序列分析获取了猪群样本中未知的6种病毒基因序列。Minakshi P[15]则从印度山羊体内鉴定到了16种血清型的蓝舌病毒。本研究通过建立一套基于病毒宏基因组学的未知病毒鉴定技术平台,快速发掘了鸭源样本中疑似坦布苏病毒的方法,可便于及时诊断和快速响应。
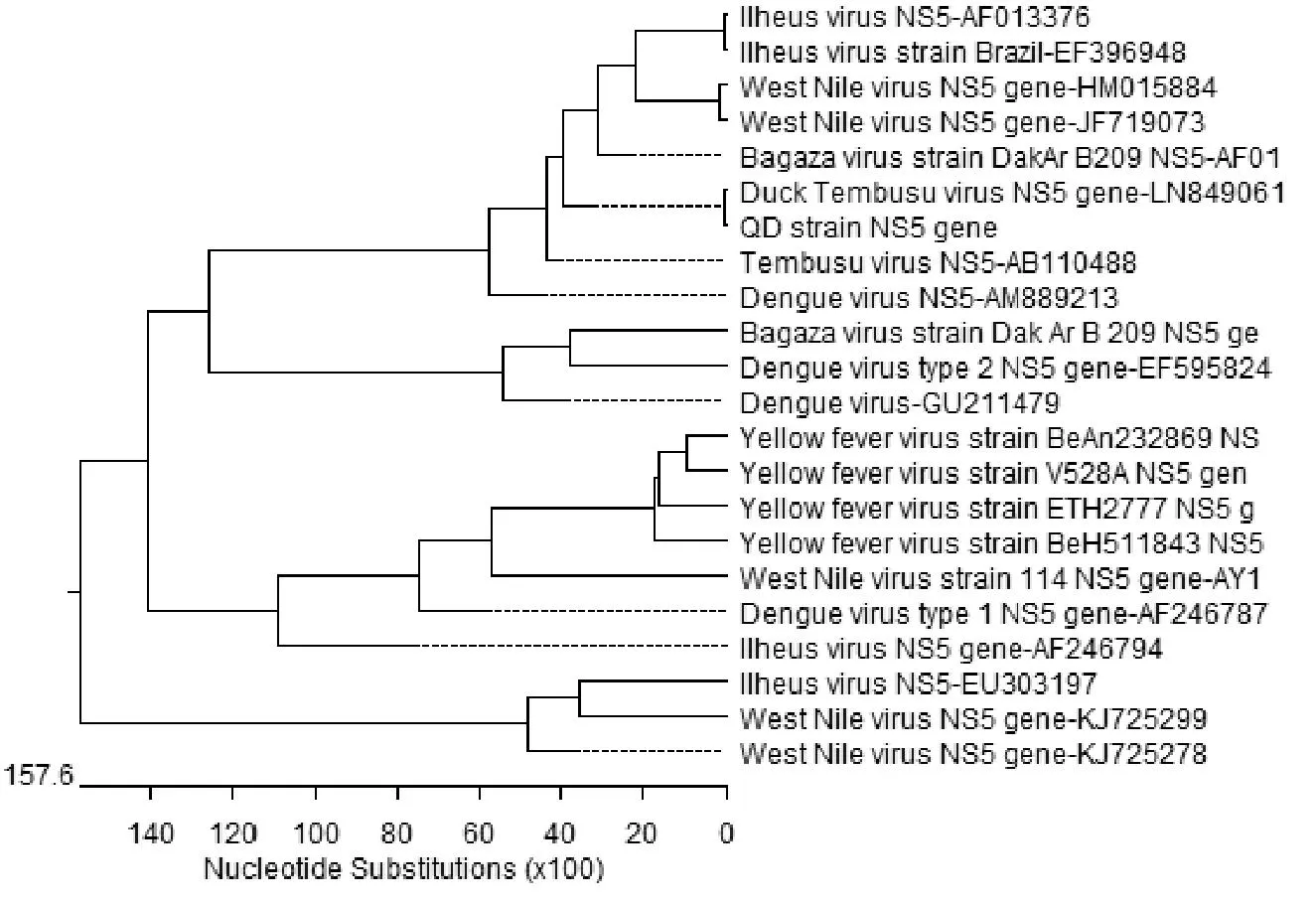
图5 NS5基因核苷酸序列比对图
在本研究中我们针对提取的RNA样本首次采用等温链位移扩增技术,高效扩增了RNA类病毒样本的核酸序列,并成功构建高质量的核酸文库。SPIA是一个强健的等温链位移扩增过程。它用一个DNA/RNA嵌合SPIA引物,DNA引物和RNA酶抑制剂在一个同质的等温实验中可提供效率很高的DNA序列扩增过程。RNA酶抑制剂在DNA/RNA异源双链的第一链5′端可降解RNA。DNA酶启动3′端的引物复制,置换了既有的上游链。新合成的链5′端RNA部分可再次被RNA酶抑制剂去除。遗弃的独特引物位点可提供新一轮的cDNA合成。整个SPIA过程由引物结合,DNA复制,第一链置换和RNA切割地等过程重复,可快速积累SPIA cDNA。
刘胜旺等[8]曾对分离自山东鸭场的病毒Du/CH/LSD/1101282株进行全基因组测序,确证了病毒的基因信息属坦布苏病毒,且与巴格扎病毒亲缘关系较近,遗传距离介于病毒种的水平上。但全基因扩增引物设计较为繁琐,且需多达10对引物的设计才最终对病毒进行了确证。在本试验中,我们采用二代高通量测序,将样品核酸进行短片段文库制备,避免了常规试验中的细菌转化、培养以及单克隆的挑选,短时间即可获得大量数据,可快速注释病原体的全部遗传信息。在本试验中,我们将本次测得的数据与其他黄病毒科成员进行同源性比对,发现全基因组的核苷酸同源性比对与近3年我国发生在鸭群中的坦布苏病毒在98%以上。选取其中的NS5基因进行核苷酸同源性比对,发现与坦布苏病毒处于同一分支上,证实该株病毒符合鸭坦布苏病毒特征,属黄病毒科、黄病毒属,蚊媒恩塔亚病毒群。
参考文献:
[1]曹贞贞,张存,黄瑜,等.鸭出血性卵巢炎的初步研究[J].中国兽医杂志,2010,46(12):3-6.
[2]Han K,Huang X,Li Y,et al.Complete genome sequence of goose Tembusu virus,isolated from jiangnan white geese in Jiangsu,China[J].Genome Announc,2013,1(2):e0023612.
[3]Huang X,Han K,Zhao D,et al.Identification and molecular characterization of a novel flavivirus isolated from geese in China[J].Res Vet Sci,2013,94(3):774-780.
[4]Su J,Li S,Hu X,et al.Duck egg-drop syndrome caused by BYD virus,a new Tembusu-related flavivirus[J].PLoS One,2011,6(3):e18106.
[5]Yun T,Ye W,Ni Z,et al.Identification and molecular characterization of a novel flavivirus isolated from Pekin ducklings in China[J].Vet Microbiol,2012,157(3-4):311-319.
[6]Zhu W,Chen J,Wei C,et al.Complete genome sequence of duck Tembusu virus, isolated from Muscovy ducks in southern China[J].J Virol,2012,86(23):131319.
[7]李玉峰.一种从鸭新分离的黄病毒研究初报[J].畜牧兽医学报,2011,42(6):885-891.
[8]刘胜旺,张玥,刘晓丽,等.一株鸭坦布苏病毒的分离鉴定及全基因组序列分析[J].东北农业大学学报,2013,44(3):72-78.
[9]马秀丽,于可响,高凤,等.鸭黄病毒BZ株的生物学特性研究[J].中国家禽,2011,33(21):12-14.
[10]Edwards R A,Rohwer F.Viral metagenomics[J].Nat Rev Microbiol,2005,3(6):504-510.
[11]Kuno G.Universal diagnostic RT-PCR protocol for arboviruses[J].J Virol Methods,1998,72(1):27-41.
[12]Ge X,Li Y,Yang X,et al.Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China[J].J Virol,2012,86(8):4620-4630.
[13]杨凡力,王意银,郑文成,等.中国部分地区蝙蝠携带病毒的宏基因组学分析[J].生物工程学报,2013,29(5):586-600.
[14]韩文,罗玉子,赵碧波,等.基于宏基因组学的猪群样本病毒探测方法的建立[J].微生物学报,2013,53(2):197-203.
[15]Minakshi P,Singh R,Ranjan K,et al.Complete genome sequence of bluetongue virus serotype 16 of goat origin from India[J].J Virol,2012,86(15):8337-8338.
Establishment of Detecting Tembusu Virus Method from Duck Source Samples Based on Metagenomics
SUN Tao1,WANG Chao2, DENG Ming-jun1,ZHENG Xiao-long1, WANG Qun1, XU Biao1
(1.ShandongEntry-exitInspectionandQuarantineBureau,Qingdao,Shandong,266002,China;2.TaianEntry-exitInspectionandQuarantineBureau,Taian,Shandong,250014,China)
Abstract:To explore the methods of high-throughput sequencing for Tembusu virus from duck source samples based on metagenomics,first of all,interference was removed through filtering and nucleic acid enzyme treatment.Then,total cDNA was prepared by using isothermal strand-displacement amplification (SPIA) technology.The purification was made by using magnetic beads and the nucleotid library building, the cDNA amplicons were sequenced by Ion Torrent,and the genome information was acquired.The results showed that 327 pmol/μL nucleic acid library was prepared from pathogenic samples through SPIA and optimizing preparation of DNA library.1 725 436 reads,about 6.2 M data,were got by Ion Torrent sequencing,and average read was 109 bp.The nucleic acid sequence of pathogen samples can note entire genome data by Blast on NCBI with the pathogen analysis software,and the similarity was 100% compared to the BYD-1 strain reference sequence occurred first time in our country.In this study,the genome of QD strain has high similarity to duck Tembusu virus occurred during nearly three years compared with the other five flaviviruses,and the genetic relationship was up to 98%.Evolutionary tree of NS5 gene analysis showed that this detected strain and duck Tembusu virus were at the same branch,conforming to the Tembusu virus's characteristics.The SPIA amplification combined with high-throughput sequencing method based on the unknown pathogens,genomic information can be synchronized to learn effectively.
Key words:metagenomics;Duck Tembusu virus;next generation sequencing
文章编号:1007-5038(2016)04-0024-06
中图分类号:S852.659.6;S852.657
文献标识码:A
作者简介:孙涛(1981-),男,山东莱芜人,硕士,主要从事动物病毒的分子生物学检测研究。*通讯作者
基金项目:国家质检总局科研项目(2013IK038)
收稿日期:2015-09-24
