
家系数据中罕见基因变异与疾病关联分析的统计方法
2016-04-21 10:49阮培峰
复旦学报(医学版) 2016年2期
阮培峰
(复旦大学计算机科学学院 上海 200433)
家系数据中罕见基因变异与疾病关联分析的统计方法
阮培峰△
(复旦大学计算机科学学院上海200433)
【摘要】目的提出一种适应家系数据的序列核关联检验(sequence kernel association test,SKAT)模型,以提高家系数据中检验罕见变异的统计模型的功效。方法提出一种适应家系数据的SKAT模型(adjusted SKAT,ADSKAT),通过对SKAT的原模型进行修改,加入表示家系结构的随机作用向量,使得家系数据中亲属相关性的影响被考虑进模型,并且得出新的检验统计量对应的概率分布。结果在家系数据中,ADSKAT不仅有效地控制了一类错误的增长,并且比现有的识别罕见变异的GWAS统计模型有着更高的统计功效。结论ADSKAT是一种在家系数据中识别与疾病关联的罕见变异的统计模型,具有广泛的应用前景。
【关键词】罕见基因变异;全基因组关联分析 (GWAS);家系数据
全基因组关联分析(genome-wide association study,GWAS)自从2005年被首次应用以来,已经发现并证实了超过2 000个与疾病或者性状关联的基因位点[1]。然而,通过GWAS识别的常见基因变异通常只能解释一小部分的疾病成因和性状遗传。而罕见变异通常指等位基因频率小于0.1%~1%的变异,在疾病和性状的遗传中可以起到非常重要的作用。常规的GWAS只对单个SNP进行检验,这样的方法对罕见基因变异的检验就显得效力不足[2]。针对这个问题,一些新方法被提出,比如Morgenthaler等在2007年[3]、Madsen等在2009年[4]以及Morris等在2010年[5]提出的负荷检验。这些方法将在事先定义的基因区域(例如基因,通路等)中所有罕见基因变异的信息压缩成一个单一的变量,进而进行检验。但这些负荷检验都存在一些缺陷,它们均预设所有的罕见基因变异的作用方向和作用程度都是一致的。然而事实上,同一区域的不同变异的作用方向和程度往往是不同的。这样就在负荷检验中导致了不必要的干扰,从而导致了检验效力的减弱。一些改进后的负荷检验,如Han等[6]、Hoffman等[7]以及Zawistowski等[8]在2010年提出的改进模型,它们不预设所有的罕见基因变异的作用方向和程度是相同的。但这些方法都引入了置换检验,用以检验统计显著性。然而,置换检验对计算能力带来了极大的挑战,尤其是在全基因组层面上进行的检验。
Wu等[9]在2011年提出了序列核关联检验(sequence kernel association test,SKAT)。这是一种基于回归分析的用以检验罕见基因变异的方法,具有应用灵活及计算效率高等优点。SKAT对于基因变异的方向及作用程度没有任何预设,同时,不同于使用置换检验的方法,SKAT可以解析地得出检验的P值。SKAT既可以对连续的表型进行关联分析检验,也可以对二分的表型进行关联分析检验。仿真数据实验和真实数据实验已经证明,SKAT比传统的负荷检验有着更高的检验功效。
在致病基因的连锁分析中,对于家系数据的研究是一种常见的研究手段[10-12]。然而,不加处理地将GWAS方法直接应用于家系数据中,比如常规的一般线性回归、Logistic回归或是SKAT,由于对家系数据中个体间的相关性的忽视,将导致统计检验中一类错误的增长。
本文中,ADSKAT通过对SKAT中假设检验的原假设进行修改,使得假设检验的原假设中包括家系结构对表型产生的影响,进而得出新的检验统计量对应的概率分布。ADSKAT的基本原理依然与SKAT一致,当不存在家系结构时,ADSKAT和SKAT是相同的。正因如此,ADSKAT和SKAT有着同样高度的灵活性。无需置换检验也使得ADSKAT有着较高的计算效率。
通过在仿真数据的实验可以看到,当没有考虑家系数据中家系结构的关系时,SKAT中出现了统计检验一类错误增长的问题。相对地,ADSKAT并不受此种问题的困扰,一类错误一直控制在正常的范围内。同时,在家系数据中,ADSKAT相对于SKAT方法也有着更高的检验功效。因而,在家系数据中检验与疾病关联的罕见基因变异时候,ADSKAT是一种更为准确高效的方法。
资 料 和 方 法
假设有n个个体,在某个遗传区域中有p个变异位点。对于第i个个体,yi表示其表型变量,Xi=(xi1,xi2,…,xim)表示协变量,Gi=(gi1,gi2,…,gip)表示在遗传区域内p个变异位点的基因型。于是,对于连续型的表型,考虑如下半参数模型:
(1)
对于二分的表型,考虑一下如下半参数Logistic模型:
(2)
其中,α0为截距,α=[α1,α2,…,αm]′是协变量的回归系数,f是一个半参数方程,由表达定理(representer therorem)[13],f可以由半正定核函数K决定:
函数K度量了第i个和第i′个个体间同一区域中p个变异的相似程度。理论上,任意一个半正定的函数K都可以用来当做这个核函数。若假设基因作用为线性,可以选择线性核函数K,则:
则式(1)和式(2)分别可以表示为:
为检验罕见变异是否和疾病关联,考虑原假设:f(Gi)=0。Liu等[14]在2008年阐明了核回归和广义线性混合模型之间的关系。令K为一个n×n的矩阵,第(i,j)个元素为K(Gi,Gj)。可以发现,f=Kγ,其中,f=[f1,f2…fn]′,则f可以被视为个体的随机效应,服从任意均值为0、方差为τK的分布。注意到τ决定了罕见变异的效应,由此,H0:f(Gi)=0与H0:τ=0等价。参考既往文献[15],检验H0:τ=0的统计量为:
另外,青海探索将缴存住房公积金事宜纳入劳动合同、聘用合同文本正式条款,为自主缴存人员依法缴存住房公积金提供有力保障。同时,建立健全不缴、欠缴住房公积金“黑名单”制度,将未按规定建立住房公积金制度或拖欠缴纳住房公积金的单位予以曝光,并纳入人民银行征信系统和政府信用信息平台,对失信主体实施联合惩戒。
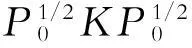
适应家系数据的SKAT改进方法(adjustedSKAT,ADSKAT)在处理家系数据时,由于SKAT没有将家系结构中个体的关联性考虑进模型中,这样的忽略会造成统计检验中一类错误的增长。为了避免这样的问题出现,一个直观的想法是,将由于家系结构而造成的影响包括进统计检验的原假设中,那么,通过拒绝原假设而得到的关联基因,就不再受到家系结构中的相关性的影响,从而避免了一类错误的增长。
首先,以性状为连续时为例,我们考虑在原半参数模型中加入表示家系结构的随机作用的项ξ:
(3)
φ为表示家系关系的亲属关系矩阵(Kinship Matrix)。
此时,表型变量yi的方差为:
此时原假设变为:
此时,使用文献[9]中相同的计算方法,统计检验量依然使用:
Q所服从的概率分布变为:

当性状为二分的时,只要将线性模型变为Logistic模型即可:
基于家系数据的负荷检验为了进一步检验ADSKAT的检验功效,我们还将ADSKAT与另一种新颖的罕见变异关联分析的方法(Family-basedrarevariantassociationtest,FARVAT)进行了比较。家系数据中,FARVAT的使用负荷检验结合方差组分检验,在家系数据中对罕见变异的检验获得了良好的效果[16]。
数据集本文采用基因分析研讨会(geneticsanalysisworkshop)发布的高血压全基因组测序数据集(http://www.gaworkshop.org/gaw18/index.html)。数据集中包含来自20个家系的849人的血压、年龄、性别以及他们的全基因组测序数据。全基因组测序中包含20万个变异位点。在其发布的由此数据得到的仿真数据中,性别、年龄等协变量保持不变,血压表型即舒张压(DBP)和收缩压(SBP)则按照真实数据中的遗传可能性进行模拟,同时,模拟的DBP和SBP也因性别、年龄的不同而异。最后,根据淋巴细胞中的基因表达情况和血压表型的关联系数,选出了表型仿真数据中的功能基因。数据中共包含200份仿真数据。
结果
一类错误ADSKAT和SKAT在不同的显著性水平下的经验一类错误(empiricaltypeIerror)结果显示(表1),如果直接将SKAT应用于存在家系结构的数据中,由于SKAT没有考虑家系中个体的相关性,在各个显著性水平下,一类错误都出现了增长的问题。相对应地,将家系结构考虑进模型的ADSKAT和FARVAT都很好地控制了一类错误的增长。

表1 不同的显著性水平下ADSKAT、SKAT和
另外,如图1所示,ADSKAT和FARVAT的P值基本服从均匀分布,但SKAT的P值分布中,越小的P值频率越高,从中也可以部分解释为什么SKAT会出现一类错误增长的结果。
检验功效我们分别对模拟数据中的SNPs的作用方向进行假设,在第一种情况下,80%的SNPs有着正效应,20%的SNPs有着负效应;在第二种情况下,50%的SNPs有着正效应,50%的SNPs有着 负效应;在第三种情况下,20%的SNPs有着正效应,80%的SNPs有着负效应。如表2所示,在3种假设情况下,通过对200个模拟数据集分别应用ADSKAT、SKAT和FARVAT,在显著水平为0.05下得到的经验检验功效都显示,ADSKAT有着最高的检验功效。可以发现,DSKAT在控制了一类错误增长的同时,检验功效高于SKAT和FARVAT。在对存在家系结构的数据进行基因组关联研究时,ADSKAT是一种更准确的检验罕见变异和疾病关联的方法。
ADSKAT and FARVAT control the type I error well while SKAT suffers the inflation of type I error.
图1ADSKAT、SKAT以及FARVAT的模拟数据试验中SNPs的P值分布
Fig 1Distribution of ADSKAT,SKAT and FARVAT’sP-values in simulation

表2 模拟数据中应用ADSKAT、SKAT和FARVAT
Under all three scenarios,ADSKAT shows the highest empirical statistical power;both ADSKAT and FARVAT perform better than SKAT as they consider the pedigree structure in their models.
讨论
本文提出了一种改进后的SKAT方法即ADSKAT,用以检验家系数据中和疾病或某些性状相关联的罕见基因变异。我们证明了ADSKAT是一种通用的进行罕见基因变异关联分析的方法。当不存在家系结构时,ADSKAT和SKAT是等价的。正因如此,ADSKAT也继承了SKAT灵活且计算效率较高的优点。
在仿真b实验中,我们证明了在相互相关的数据中直接应用SKAT会导致一类错误的增长。这时,如果要使用SKAT,只能将相关的数据删除,只对独立的数据进行关联分析。然而,这样显然会无谓地失去许多有用的信息,同时样本量的减少也会导致检验功效的降低。相对地,应用ADSKAT并不需要减少样本量。ADSKAT在控制一类错误的增长的同时,也有着较高的检验功效。可见,在检验罕见基因变异与疾病的关联时,ADSKAT是一种更好的选择。
尽管ADSKAT是为了进行罕见基因变异关联分析而设计的,但它依然可以用来检验常见基因变异与疾病的关联,或者同时进行常见/罕见基因变异的关联分析。对于常见基因变异,我们可以将之视为(3)式中的固定效应,或者与罕见变异一起作为随机效应来检验。
通过对权重的选择,可以进一步提高ADSKAT检验的功效。
参考文献
[1]VISSCHER PM,BROWN MA,MC CARTHY MI,etal.Five years of GWAS discovery[J].AmJHumGenet,2012,90(1):7-24.
[2]SCHORK NJ,MURRAY SS,FRAZER KA,etal.Common vs.rare allele hypotheses for complex diseases[J].CurrOpinGenetDev, 2009,19(3):212-219.
[3]MORGENTHALER S,THILLY WG.A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases:a cohort allelic sums test (CAST)[J].MutatRes, 2007,615(1-2):28-56.
[4]MADSEN BE,BROWNING SR.A groupwise association test for rare mutations using a weighted sum statistic[J].PLoSGenet,2009,5(2):e1000384.
[5]MORRIS AP,ZEGGINI E.An evaluation of statistical approaches to rare variant analysis in genetic association studies[J].GenetEpidemiol,2010,34(2):188-193.
[6]HAN F,PAN W.A data-adaptive sum test for disease association with multiple common or rare variants[J].HumHered,2010,70(1):42-54.
[7]HOFFMANN TJ,MARINI NJ,WITTE JS.Comprehensive approach to analyzing rare genetic variants[J].PLoSOne,2010,5(11):e13584.
[8]ZAWISTOWSKI M,GOPALAKRISHNAN S,DING J,etal.Extending rare-variant testing strategies:analysis of noncoding sequence and imputed genotypes[J].AmJHumGenet,2010,87(5):604-617.
[9]WU MC,LEE S,CAI T,etal.Rare-variant association testing for sequencing data with the sequence kernel association test[J].AmJHumGenet,2011,89(1):82-93.
[10]FALK CT,RUBINSTEIN P.Haplotype relative risks:an easy reliable way to construct a proper control sample for risk calculations[J].AnnHumGenet,1987,51(3):227-233.
[11]OTT J.Statistical properties of the haplotype relative risk[J].GenetEpidemiol,1989,6(1):127-130.
[12]TERWILLIGER JD,OTT J.A haplotype-based haplotype relative risk’ approach to detecting allelic associations[J].HumHered,1992,42(6):337-346.
[13]KIMELDORF G,WAHBA G.Some results on Tchebycheffian spline functions[J].JMathAnalAppl,1971,33(1):82-95.
[14]LIU D,GHOSH D,LIN X.Estimation and testing for the effect of a genetic pathway on a disease outcome using logistic kernel machine regression via logistic mixed models[J].BMCBioinformatics,2008,9(1):292.
[15]ZHANG D,LIN X.Hypothesis testing in semiparametric additive mixed models[J].Biostatistics,2003,4(1):57-74.
[16]CHOI S,LEE S,CICHON S,etal.FARVAT:a family-based rare variant association test[J].Bioinformatics, 2014,30(22):3197-3205.
A statistical method for rare variants association studies in pedigree data
RUAN Pei-feng△
(SchoolofComputerScience,FudanUniveristy,Shanghai200433,China)
【Abstract】ObjectiveTo propose an adjusted sequence kernel association test (SKAT) model in order to identify rare variants for pedigree data which has higher statistical power.MethodsIn this paper,we proposed a SKAT model fitting pedigree data (ADSKAT).The SKAT model was modified by adding a random effect vector of pedigree structure into the model.Thus the influence of kinship correlation was taken into consideration in the new model.A new distribution of test statistics was defined.ResultsSimulations demonstrated that ADSKAT well controlled the inflation of type I error and achieved better statistical power than the existed mainstream methods for identifying disease-related rare variants.ConclusionsADSKAT has broad application prospects in the fields of identifying disease related rare variants in pedigree data.
【Key words】rare variants;genome-wide association study;pedigree data
(收稿日期:2015-10-12;编辑:张秀峰)
【中图分类号】TP399,R181.2+3
【文献标识码】A
doi:10.3969/j.issn.1672-8467.2016.02.018
△Corresponding authorE-mail:pruan12@fudan.edu.cn
