
HPLC测定注射用奥沙利铂的含量及有关物质
2016-04-19 01:46陈传喜杨志勇王继平王大荣
安徽医药 2016年3期
关键词:含量
陈传喜,杨志勇,袁 红,王继平,王大荣
(黄冈市中心医院肿瘤科,湖北 黄冈 438000)
◇药物分析◇
HPLC测定注射用奥沙利铂的含量及有关物质
陈传喜,杨志勇,袁红,王继平,王大荣
(黄冈市中心医院肿瘤科,湖北 黄冈438000)
摘要:目的建立注射用奥沙利铂含量及有关物质的HPLC方法。方法含量和有关物质测定以十八烷基硅烷键合硅胶作为填充剂Zorbax Rx-C(18)(4.6 mm×250 mm)5 μm,以磷酸溶液(取10%磷酸溶液0.6 mL,加水稀释至1 000 mL,用氢氧化钠溶液或磷酸调节pH值至3.0)—乙腈=99∶1为流动相,流速0.8 mL·min(-1),检测波长209 nm,进样量20 μL。 结果奥沙利铂在6~16×10(-3) g·L(-1)浓度范围内线性关系良好,检测限为0.02%,回收率为100.55%(RSD=0.46%),测定样品含量为99.8%,有关物质均小于0.5%。结论该方法快速,专属性强,灵敏度高,重现性好,可作为注射用奥沙利铂的含量及有关物质的检测方法。
关键词:注射用奥沙利铂;HPLC法;含量;有关物质
注射用奥沙利铂是第三代铂类抗癌药物,临床用于治疗晚期结、直肠癌,对顺铂耐药的肿瘤细胞也有一定作用,且毒副作用比顺铂小[1],与传统化疗药物或紫杉类药物间不具交叉耐药性[2-3]。注射用奥沙利铂(商品名:乐沙定RR)由瑞士Debiopharm公司研制开发生产,规格为50 mg,于1996年首次在法国上市,现已经在欧盟、美国及我国等多个国家和地区上市多年,临床应用广泛。铂类药物是晚期癌症的首选方案,但是早期开发的顺铂和卡铂具有明显的交叉耐药性。奥沙利铂含有DACH基团空间位阻作用较强,作用机制与顺铂类似,与其他抗癌药物联合使用无交叉耐药性,能有效避免上述缺点。该药的分子式为C18H14N2O4Pt,分子量397.29,化学名为 (1R-反式)-(1,2-环己二胺-N,N’)[草酸(2-)-O,O’] 合铂。美国药典收载了注射用奥沙利铂的质量标准[4],中国药典、英国药典等均未收载注射用奥沙利铂的质量标准,国内原有质量标准WS1-(X-090)-2003Z是为奥沙利铂的无菌冻干品或奥沙利铂与乳糖的无菌冻干品制定,但研究发现此方法不能将有关物质中的单杂进行控制。本文通过对现有HPLC的条件进行优化,得到快速、准确的测定注射用奥沙利铂含量及有关物质的方法。
1仪器与试药
Agilent 1260型高效液相色谱仪,配紫外检测器(Agilent)及安捷伦色谱工作站,电子分析天平(AE240 梅特勒-托利多仪器有限公司),HDC-S超声仪(济宁亨达超声设备有限公司)。色谱纯乙腈(色谱纯,TEDIA公司生产),纯化水,氢氧化钠(分析纯,天津市科密欧化学试剂有限公司),磷酸(分析纯,国药集团化学试剂有限公司)。奥沙利铂标准品(中国药品生物制品检定所,批号100584-201408),注射用奥沙利铂(江苏恒瑞医药股份有限公司,批号:14082247,14082547,14092147,规格50 mg)。
2方法和结果
2.1色谱条件 色谱柱:Zorbax Rx-C18(4.6 mm×150 mm,5 μm);流动相:磷酸溶液(取10%磷酸溶液0.6 mL,加水稀释至1 000 mL,用氢氧化钠溶液或磷酸调节pH值至3.0)∶乙腈=99∶1为流动相,流速0.8 mL·min-1,检测波长209 nm,进样量20 μL;柱温:30℃。
2.2溶液配制
2.2.1含量测定对照品溶液精密称取经105℃干燥至恒重的奥沙利铂对照品25 mg,置250 mL容量瓶中,加适量纯化水溶解后,定容至刻度,摇匀,配制成浓度为0.1 g·L-1的对照品溶液,备用。
2.2.2含量测定供试品溶液取供试品注射用奥沙利铂适量(相当于奥沙利铂25 mg),置250 mL容量瓶中,加适量纯化水溶解后,定容至刻度,摇匀,配制成浓度为0.1 g·L-1的供试品溶液,备用。
2.2.3有关物质测定供试品溶液称取“2.2.2”项中注射用奥沙利铂的粉末适量,精密称定,置于50 mL的容量瓶中,加入适量蒸馏水溶解,补加蒸馏水定容,摇匀。配制成1 mL中含有约0.01 mg奥沙利铂的溶液。
2.2.4有关物质对照溶液精密量取“2.2.3”项中的有关物质供试品溶液1 mL,置于100 mL的量瓶中,加入纯化水定容至刻度,摇匀。
2.2.5阴性对照溶液称取甘露醇100 mg、置于100 mL量瓶中,加水稀释至刻度,滤过,取适量,稀释与样品相同倍数,即得空白辅料溶液。
2.3方法学考察
2.3.1专属性试验取“2.2.1”项下奥沙利铂对照品溶液1 mL,置于100 mL的容量瓶中,加入纯化水定容至刻度,充分振摇溶解,精密量取 20 μL进色谱仪,按照上述色谱条件测定,记录色谱图。按照上述方法分别取“2.2.2”项下含量供试品溶液、 “2.2.3”项有关物质供试品溶液、“2.2.4”项有关物质对照溶液及“2.2.5”空白辅料溶液各1 mL,稀释至同等浓度,进色谱仪测定,记录色谱图,结果辅料峰未干扰测定(图1)。
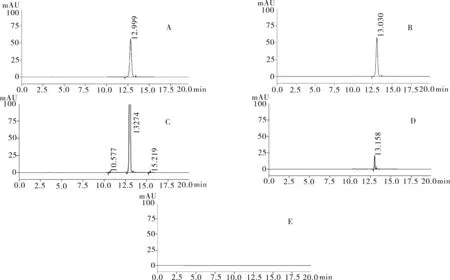
图1 HPLC色谱图
注:A.含量测定对照品溶液;B.含量测定供试品溶液;C.有关物质测定供试品溶液;D.有关物质对照溶液;E.阴性对照溶液。
再分别取注射用奥沙利铂适量,研细,精密称量粉末(约相当于主药10 mg)进行如下测试:(1)酸破坏:将精密称定后的粉末置具塞试管中,加入0.1 mol·L-1盐酸溶液25 mL,摇匀,置于100℃水浴锅中4 h,放冷。加入0.1 mol·L-1氢氧化钠中和并转移至100 mL的容量瓶中,用流动相稀释至刻度,配制成浓度为0.1 g·L-1的溶液,摇匀,过滤。(2)碱破坏:将精密称定后的粉末置具塞试管中,用0.1 mol·L-1氢氧化钠溶液25 mL溶解,摇匀,置于100℃水浴锅中4 h,放冷。加入0.1 mol·L-1盐酸中和并转移至100 mL的容量瓶中,用流动相稀释,配制成浓度为0.1 g·L-1的溶液,摇匀,过滤。(3)氧化破坏:将精密称定后的粉末置具塞试管中,加入30%过氧化氢溶液10 mL,摇匀,放置12 h,加水稀释至刻度,配制成浓度为0.1 g·L-1的溶液,摇匀,过滤。(4)加热破坏:将精密称定后的粉末置具塞试管中,加水稀释,配制成浓度0.1 g·L-1的溶液25 mL,置于100℃水浴锅中4 h,放冷,过滤。(5)光破坏:将精密成定后的粉末置具塞试管中,加水稀释,配置成0.1 g·L-1溶液25 mL,置于强光(5 000 Lx)下照射12 h后,放冷。按照上述色谱条件进行测定,记录色谱图。结果显示样品在经过酸、碱、氧化、加热、光照破坏后主成分与各降解产物可以达到很好的分离(图2)。
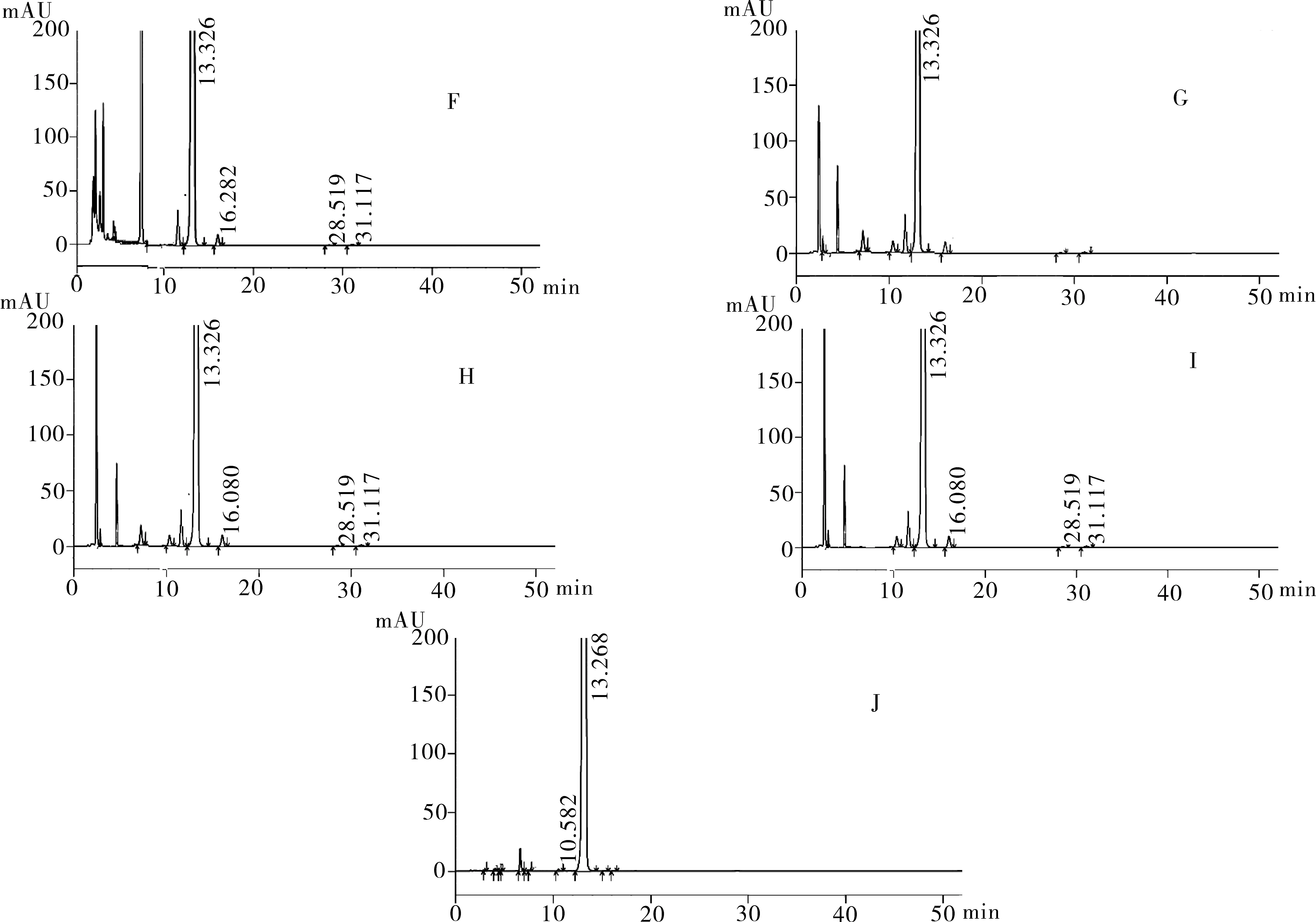
图2 降解产物测试HPLC图
注:F.样品酸破坏图谱;G.样品碱破坏图谱;H.样品氧化破坏图谱;I.样品加热破坏图谱;J.样品光照破坏图谱。
2.3.2系统适用性试验分别精密量取“2.2.1”项中奥沙利铂对照品溶液、“2.2.2”注射用奥沙利铂供试品溶液及“2.2.5”项阴性对照溶液20 μL,按照上述色谱条件进样,色谱柱的理论塔板数按照奥沙利铂计算不得少于2 000,奥沙利铂与相邻杂质峰的分离度不低于2.0。
2.3.3检测限测定取“2.2.1”项中配制的奥沙利铂对照溶液,稀释至50×10-3g·L-1,按照上述色谱条件进样,根据信噪比法进行试验,得到的结果:最小检出量为4×10-6mg(S/N=3),检出限为0.02%。
2.3.4线性试验精密称量奥沙利铂对照品25 mg,置于25 mL的容量瓶制备成浓度为1.0 g·L-1的溶液,依次精密吸取0.6,0.8,1.0,1.2,1.4,1.6 mL,分别置于100 mL容量瓶中,用流动相稀释至刻度,摇匀,得到浓度依次为6.00,8.01,10.01,12.02,14.02,16.02×10-3g·L-1浓度的溶液,按照上述色谱条件取20 μL依次进样,记录色谱图。以奥沙利铂峰面积Y为纵坐标,以进样浓度X为横坐标,得到线性回归方程为:Y=0.170 1X+0.141 4,r=0.999 8,表明奥沙利铂在6.00~16.02×10-3g·L-1浓度范围内呈现良好的线性关系,结果见图3。

图3 奥沙利铂线性关系图
2.3.5精密度及重复性试验取“2.2.1”项下奥沙利铂对照溶液,按照上述色谱条件进样,重复测定6次,记录奥沙利铂的峰面积,计算相标准偏差RSD=0.39%,说明仪器精密度良好。此外,取14082247批次的注射用奥沙利铂,精密称量适量的粉末(相当于主药50 mg),加水稀释,配制成浓度为1 g·L-1供试品溶液6份,按照上述色谱条件在同一高效液相色谱仪上分别进样,记录奥沙利铂面积,其含量平均值为99.78%,相对标准偏差RSD=0.42%,显示重现性良好。
2.3.6回收率试验按照处方比例称量适量的辅料,(相当于1瓶制剂的辅料量),分别加入处方量的80%,100%,120%的奥沙利铂的对照品,加水溶解并稀释至刻度,摇匀,每个含量的溶液分别取3份1 mL加流动相稀释至10 mL作为供试品溶液。精密量取20 μL溶液,按照上述色谱条件进液相仪,记录图谱,结果见表1。计算得平均回收率为100.55%,RSD=0.46%。
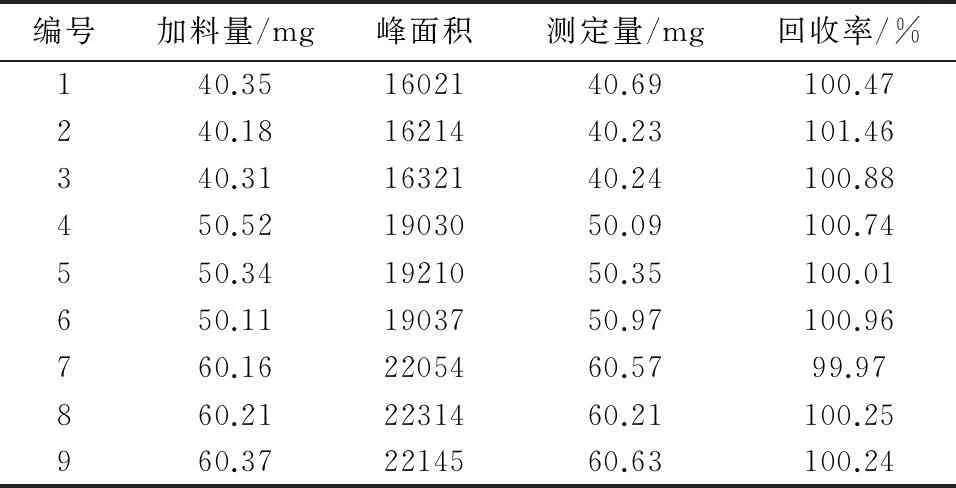
表1 加样回收率数据
2.3.7溶液稳定性试验取供试品溶液,分别在0、6、12、18、24 h精密吸取20 μL进样,记录色谱图,得出峰面积分别为19021、19214、19321、19330、19610,平均峰面积19226,计算其相对标准偏差RSD为0.65%,说明奥沙利铂在24 h内溶液稳定。
2.4样品测定
2.4.1有关物质测定取上市品注射用奥沙利铂1瓶,精密称量适量粉末(相当于奥沙利铂50 mg),置于50 mL的容量瓶中,加水振荡溶解,补加水稀释至刻度,摇匀,过滤,滤液作为供试品。取“2.2.1”项中奥沙利铂对照溶液,稀释与供试品同等浓度作为对照溶液。精密量取供试品和对照品溶液20 μL进高效液相色谱仪,记录色谱图。根据面积归一化法计算有关物质的峰面积。按照上述方法分别测定3批上市品(批号:14082247,14082547,14092147)中有关物质测定结果分别为0.35%,0.33%,0.36%。
2.4.2含量测定取上市品注射用奥沙利铂1瓶,精密称量适量粉末(约相当于主药10 mg),置于100 mL的容量瓶中,加水溶解,补加水稀释至刻度,摇匀,作为供试品。取“2.2.1”项中奥沙利铂对照溶液稀释与供试品同等浓度作为对照溶液。精密量取20 μL对照品与供试品溶液,进高效液相色谱仪,记录图谱。按照外标法分别测定3批上市品(批号:14082247,14082547,14092147)中含量分别为99.8%,99.9%,99.8%。
3讨论
在对上市品有关物质及含量方法学的研究过程中[5-6],按照国内药品标准[7]研究发现此条件不能有效的将有关物质进行分离,分离度不足1.5,不能作为检测有关物质的条件进行测定。
本研究通过调整流动相的比例和流速,尝试使用Alltima-C18、Zorbax Rx-C18、HP-35-C18柱在分离度、理论塔板数、拖尾因子等方面综合考量,Zorbax Rx-C18柱具有能有效分离奥沙利铂的有关物质草酸及其他杂质,出峰时间合理。另外通过专属性研究酸、碱、高温、氧化破坏的杂质及对照品最大的吸收峰均在209 nm,此波长可以将主成分及有关物质较为灵敏的检测出。
通过测定的结果显示在以下条件:磷酸溶液(1 mL磷酸,加水至1 000 mL即得)—乙腈(99∶1);流速:0.8 mL·min-1;检测波长:209 nm;进样量:20 μL;柱温:30℃,主峰和辅料、有关物质达到基线分离要求,色谱峰形对称符合检测要求。
参考文献:
[1]王彦美,韩杰,周邵兵,等.铂抗癌药物研究进展[J].合成化学,2003,11(1):27-31.
[2]张文.晚期结直肠癌的化疗进展[J].中国癌症杂志,2004,14(4):380.
[3]杨柳青,秦叔逵.第3代铂类药物洛铂的研究新进展[J].临床肿瘤学杂志,2009,14(12):1134-1139.
[4]United States Pharmacopoeia(31)[S],2015:2696.
[5]徐静,张伟明,李健和,等.奥沙利铂静脉输液的有关物质检查与含量测定[J].中国药业,2013,22(15):48-50.
[6]闫占宽,李语如,宋晓光,等.高效液相色谱法测定奥沙利铂注射液中奥沙利铂的含量和有关物质[J].东华理工大学学报,2010,33(3):290-292.
[7]国家药品标准. 注射用奥沙利铂药品注册标准[S],2003.
Determination of content and related substances of Oxaliplatin for Injection by HPLC
CHEN Chuan-xi,YANG Zhi-yong,YUAN Hong,et al
(DepartmentofOncology,HuanggangCentralHospital,Hubei438000,China))
Abstract:ObjectiveTo establish an HPLC method to determine the oxaliplatin content and the related substances of oxaliplatin for injection. Methods The chromatographic separation was performed on Zorbax Rx-C(18)(4.6 mm×250 mm,5 μm)that was made by alkyl-bonded silica gels,phosphoric acid solution(10% phosphoric acid solution 0.6 mL,diluted with water to 1 000 mL,pH adjusted to 3.0 with sodium hydroxide solution or phosphoric acid)- acetonitrile=99∶1; the flow rate was 0.8 mL·min(-1);the detection wavelength was 209 nm; the sample size was 20 μL.Results The linear of oxaliplatin was 6~16×10(-3) g·L(-1). The limits of detection was 0.02%. The average recovery was 100.55%(RSD=0.46%). Determination of the sample average content was 99.8%,and the related substance was less than 0.5%.Conclusions The method is rapid,specific,sensitive,and well repeatable,which can be used to determine the content of oxaliplatin for injection and its related substances.
Key words:oxaliplatin for injection;HPLC;content;related substance
(收稿日期:2015-10-23,修回日期:2016-01-18)
doi:10.3969/j.issn.1009-6469.2016.03.012
猜你喜欢
山东冶金(2022年3期)2022-07-19
玩具世界(2022年1期)2022-06-05
氯碱工业(2021年6期)2021-12-25
四川蚕业(2021年2期)2021-03-09
天津医科大学学报(2021年1期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
四川冶金(2019年4期)2019-11-18
中成药(2017年10期)2017-11-16
天然产物研究与开发(2016年6期)2016-06-05
中国资源综合利用(2016年10期)2016-01-22
