
气相色谱法测定汽油馏分中微量小分子含氧化合物
2016-04-12 02:09李长秀王亚敏
石油炼制与化工 2016年8期
李长秀,王亚敏,金 珂
(中国石化石油化工科学研究院,北京 100083)
气相色谱法测定汽油馏分中微量小分子含氧化合物
李长秀,王亚敏,金 珂
(中国石化石油化工科学研究院,北京 100083)
建立了一套带有反吹组件和微流控中心切割组件及3根毛细管色谱柱的色谱系统,可以用于测定汽油中微量小分子含氧化合物的含量,通过载气压力切换的方式也可用于直接测定微反产物汽油馏分中微量小分子含氧化合物的含量,样品中所含的大量烃类组分不干扰测定结果。通过载气压力的切换使得只有沸点小于正十一烷的组分全部从2 m长的预切柱流出进入与之相连的非极性色谱柱,其余的重组分被反吹出色谱系统。沸点小于正十一烷的组分进入一根30 m长的非极性色谱柱后,通过中心切割组件电磁阀的切换仅使沸点小于2-己酮的组分进入与之相连的10 m长强极性OxyPLOT色谱柱,实现烃类组分和含氧化合物的分离,并通过火焰离子化检测器检测,采用外标法定量。该方法可以定量检测汽油馏分中微量的C1~C4醇、C2~C5醛、C3~C6酮、甲基叔丁基醚、乙基叔丁基醚和甲基叔戊基醚的含量,单组分的检测限为0.5~2.0 μg/g,各组分测定的加标回收率基本在80%~120%之间,测定结果的相对标准偏差在2%~5%的范围内。对催化裂化和催化裂解工艺的汽油产品中含氧化合物的分析测定结果显示,汽油中的小分子含氧化合物以酮类化合物为主,同时含有少量的醛、醇、醚类组分。
汽油 含氧化合物 中心切割 气相色谱
石脑油作为蒸汽裂解生产乙烯的原料,如果其中含氧化合物的含量较高,会使生成的乙烯中含氧化合物杂质含量高而影响乙烯的质量,因此需要检测石脑油中微量含氧化合物的含量。另外,在催化裂解多产丙烯的工艺过程中,丙烯产物中的微量含氧化合物杂质同样会影响丙烯产品的质量,为探索含氧化合物在工艺过程中的生成及分布规律,除对气相产物中的甲醇、丙酮等含氧化合物的含量进行测定外,同时需测定相应的微反产物及汽油馏分中微量甲醇、丙酮等含氧化合物的含量。因此建立适合于汽油馏分微量含氧化合物测定的分析方法,对工艺研究有重要意义。
目前国内用于测定成品汽油中甲醇等含氧化合物含量的方法一般为SH/T 0663[1](等效ASTM D4815)方法或选择性的氧检测器(O-FID)测定法[2],这些方法对于一般含氧组分的最低检测限为0.1%。由于汽油的组成相当复杂,其中有些含氧化合物的含量很低,低至几μg/g,这些常规方法很难将微量的含氧化合物与烃类组分分离。微流控中心切割技术是近几年发展起来的一种新的色谱技术[3-4],采用全程电子流量控制的压力切换,可以实现毛细管色谱柱之间精确到目标组分的切换。目前研究人员采用微流控中心切割技术测定高纯度乙苯中微量二甲苯含量[5]以及苯乙烯中的微量苯含量[6],获得了很好的效果。选择合适的双柱系统完全可以分离目标组分,实现微量甲醇等含氧化合物的分析测定。
本课题建立采用带有反吹组件和中心切割(Deans switch)组件及3根色谱柱的分析系统,首先通过压力切换的方式,仅使沸点小于正十一烷的组分通过预切柱进入与之相连的非极性色谱柱,在非极性的色谱柱上,组分按照沸点顺序分离,将沸点小于2-己酮的组分切割至对含氧化合物有特殊保留的色谱柱上,将烃类组分与含氧化合物分离,消除大量的烃类组分对微量含氧化合物测定的影响;并将所建立的方法用于测定油样中微量C1~C4醇、C2~C5醛、C3~C6酮、甲基叔丁基醚、乙基叔丁基醚、甲基叔戊基醚的含量。
1 实 验
1.1 仪器和试剂
仪器:Agilent7890型气相色谱仪,配有中心切割系统、反吹组件、分流/不分流进样口和双氢火焰离子化检测器(FID)。色谱柱:HP-5毛细管色谱柱(2 m×0.32 mm×0.25 μm);DB-1毛细管色谱柱(30 m×0.32 mm×1.0 μm);OxyPLOT毛细管色谱柱(10 m×0.53 mm×40 μm)。
试剂:甲醇,乙醇,异丙醇,正丙醇,异丁醇,仲丁醇,叔丁醇,乙醛,正丙醛,异丙醛,正丁醛,异丁醛,正戊醛,异戊醛,丙酮,丁酮,2-戊酮,3-戊酮,2-己酮,3-己酮,2-甲基-3-丁酮,2-甲基-3-戊酮,3-甲基-2-戊酮,4-甲基-2-戊酮,甲基叔丁基醚,乙基叔丁基醚,二异丙醚,甲基叔戊基醚,正庚烷。以上试剂均为分析纯。
1.2 色谱系统的建立
色谱系统的连接示意见图1。该系统包含3根色谱柱:预切柱为一根2 m长的非极性或弱极性毛细管色谱柱,一端与分流进样口相连接,一端连接于反吹组件(三通)的入口;色谱柱1为一根30 m长的非极性柱,一端与反吹组件的出口相连,另一端与中心切割组件的入口相连;中心切割组件的两个出口分别与色谱柱2(强极性OxyPLOT色谱柱)以及与该色谱柱阻力相匹配的一段弹性石英阻尼柱相连接,OxyPLOT色谱柱和弹性石英柱管的出口分别连接至两个火焰离子化检测器。
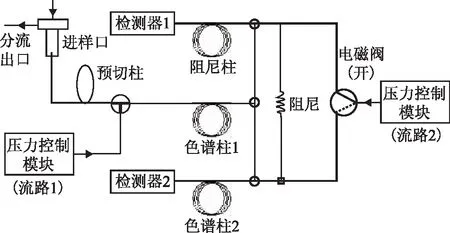
图1 微反产物含氧化合物测定系统的连接示意
当试样通过分流进样口注入色谱系统后,试样中的组分首先进入与进样口相连的预切柱中。在预切柱上,组分按照沸点的顺序分离。当沸点小于正十一烷的组分全部从预切柱流出进入与之相连的非极性色谱柱后,改变进样口的压力,使之小于由压力控制模块提供的辅助气流路1的压力,使通过预切柱的载气流动方向改变,将预切柱中沸点大于正十一烷的重组分通过分流出口反吹出色谱系统。
在进样状态下,设定与中心切割组件相连接的电磁阀的位置为开,分析开始后,使进入与预切柱相连的色谱柱1的组分流出后,进入与之相连的对含氧化合物有特殊保留能力的强极性色谱柱2中。当2-己酮从色谱柱1流出进入色谱柱2后,改变电磁阀的位置,使其余的组分不进入强极性色谱柱,而是通过阻尼柱进入火焰离子化检测器1检测。进入强极性色谱柱的组分在该色谱柱上实现烃类组分和含氧化合物的分离,并通过另一个火焰离子化检测器2检测。
采用双柱压力切换方式测定石脑油中微量含氧化合物含量的研究已有文献报道[7],但以中心切割方式连接的双柱系统用于测定含重组分的微反产物时,重组分进入非极性色谱柱后很难全部从检测器流出。直接采用反吹的方式连接非极性柱和强极性柱,尽管也可以实现重组分的反吹,但长期来看,重组分进入非极性柱后会有累积,污染非极性色谱柱,进而对微量组分的测定产生一定的影响。
该系统在双柱前增加了一根短的预柱来进行重组分反吹,其优点是既满足了将重组分反吹出色谱系统的要求,又保留了中心切割的功能。在实际应用中,当某些组分较难定性时,可以根据色谱柱1的分离情况,通过设定电磁阀的开关时间,将色谱柱1上的某个色谱峰切割至色谱柱2上进一步分离,有效消除其它组分的干扰。
1.3 实验条件
进样口:250 ℃,分流模式,分流比为10∶1;载气为氮气,稳压方式,进样口的初始压力为72.26 kPa,保持一定的时间,然后以700 kPa/min的速率降低至56 kPa;压力控制模块的压力分别为68.95 kPa(流路1)和29.51 kPa(流路2);色谱柱初温为100 ℃,保持5 min,升温速率为5 ℃/min,终温为210 ℃,保持8 min;检测器:双FID,温度为280 ℃,氢气流量为30 mL/min,空气流量为350 mL/min,氮气(补偿气)流量为25 mL/min;电磁阀在进样后0.1 min时打开,4.2 min时关闭;进样体积为1 μL。
2 结果和讨论
2.1 预切柱反吹时间的确定
为反吹重组分,进样口初始压力保持一定时间(t0)后,以700 kPa/min的速率降低至56 kPa。压力控制模块载气1(流路1)连接至2 m预柱的出口,并提供稳定的压力68.95 kPa,在时间t0后,由于预切柱出口压力大于进样口的压力,载气将由预切柱的出口反向流经预切柱后从分流出口流出,从而将时间t0后保留在预切柱上的重组分通过分流出口反吹出色谱系统。在不同预切柱反吹时间下,某石脑油样品通过色谱柱1和阻尼柱经检测器1检测得到的色谱图见图2。由图2可见,当t0为0.7 min时,刚好所有正十一烷之前的组分进入色谱柱1。因此,确定预切柱的反吹时间为0.7 min。

图2 预切柱反吹时间的确定1—正癸烷;2—正十一烷;3—正十二烷
2.2 电磁阀切换时间的确定
在确定的色谱条件下,由于在非极性色谱柱上,C1~C4醇、C2~C5醛、C3~C6酮、甲基叔丁基醚、乙基叔丁基醚、甲基叔戊基醚组分中,2-己酮最后一个出峰,因此,分析测定含约100 μg/g 2-己酮的正庚烷溶液,记录色谱图,确定2-己酮全部流出色谱柱所需的时间,结果为4.2 min,在此时间点关闭电磁阀,使2-己酮之后流出非极性色谱柱的组分不进入极性色谱柱中,而是通过阻尼柱进入检测器中检测。
2.3 定性和定量分析
组分的定性全部根据标样的保留时间确定,并通过GC/MS对催化裂化汽油中含量较高的主要组分进行确认。以正庚烷为溶剂配制的混合定性标样的色谱图见图3,标样中含氧化合物组分的保留时间及沸点见表1。

图3 含氧化合物混合标样的定性色谱图
组分的定量分析采用外标法进行。分别称量约80 mg(精确至0.1 mg)的C1~C4醇、C2~C5醛、C3~C6酮、甲基叔丁基醚、二异丙醚和甲基叔戊基醚,加入10 g正庚烷,配制成单组分质量分数约8 000 μg/g的混合醇、醛、酮、醚标准溶液。按照称量法用正庚烷逐级稀释分别得到约2,10,20,80,200 μg/g的含氧化合物标样溶液。按照选定条件分析标样溶液,对每个组分以含量为横坐标、色谱峰面积为纵坐标,绘制各组分的定量校正曲线,各组分校正曲线的相关系数R2均大于0.99,在质量分数2~200 μg/g范围内组分具有线性响应。

表1 标样中含氧化合物组分的保留时间及沸点
样品测定时,根据待测样品中组分的峰面积,通过标准曲线可以求得组分的含量。对于标样中不含的异构醇、酮组分,以同碳数和类型组分的标准曲线进行计算。某典型微反产物样品的色谱图见图4。
各组分定量的标准曲线方程可用式(1)表示。
A=aiC+bi
(1)
式中:A为组分色谱峰面积;C为组分质量分数,μg/g;ai、bi分别为不同组分标准曲线方程的系数。
待测试样中含氧化合物组分含量按式(2)计算。
(2)
式中:Ci为待测组分质量分数,μg/g;Ai为待测组分色谱峰面积;ai、bi分别为不同组分标准曲线方程的系数;ρ0为配制标样所用正庚烷的密度,0.684 g/cm3;ρs为试样的密度。

图4 某典型微反产物样品的色谱图1—甲基叔丁基醚;2—丙醛;3—异丁醛;4—正丁醛;5—丙酮;6—异戊醛;7—正戊醛;8—丁酮;9—3-甲基-2-丁酮;10—2-戊酮;11—3,3-二甲基-2-丁酮;12—2-甲基-3-戊酮;13—3-甲基-2-戊酮+异丁醇+仲丁醇+叔丁醇;14—3-己酮+4-甲基-2-戊酮+正丁醇;15—2-己酮;16—轻烃组分;17—重烃组分
2.4 方法的重复性和加标回收率
取一个典型的石脑油样品,向其中加入约50 μg/g的各种含氧化合物,在选定的条件下分析测定6次,计算加标回收率及相对标准偏差,结果见表2。对于一般的微量分析,加标回收率一般应达到80%~120%。由表2可知,除乙醛的加标回收率为127%外,其它组分的加标回收率均在80%~120%之间。乙醛加标回收率偏高的原因,可能是由于乙醛的沸点较低,常温下很容易汽化,在标样配制测定标准曲线时会引入称量误差,乙醛组分标准曲线的相关系数R2为0.990 7,是所有组分标准曲线中最低的,说明其测定结果的准确度较低。
由表2还可以看出,各组分测定结果的相对标准偏差在2%~5%的范围内,说明测定结果的重复性较好,可以满足一般分析的精密度要求。
由于本方法的定量采用了外标法,为考察定量标准曲线的稳定性,对上述实验样品在不同日期的测定结果进行比较,计算其平均加标回收率和相对标准偏差,结果列于表3。由表3可知,不同日期测定结果的相对标准偏差为1.9%~5.4%,证明本方法的定量分析结果有很高的精密度和稳定性。

表2 测定结果的加标回收率和相对标准偏差

表3 不同日期测定结果的加标回收率和相对标准偏差
2.5 方法的检测限
配制各组分质量分数约为2.0 μg/g的标样,逐渐稀释标样,在选定条件下进行分析,确定各组分的检测限。当信噪比为3时,各组分的检测限见表4。由表4可知:C1~C3醇的检测限较高,为1.0~2.0 μg/g,这是由于C1~C3醇的极性较强,存在一定的吸附作用;乙醛和丙醛的检测限分别为2.0 μg/g和1.0 μg/g,其它含氧化合物的检测限均为0.5 μg/g。
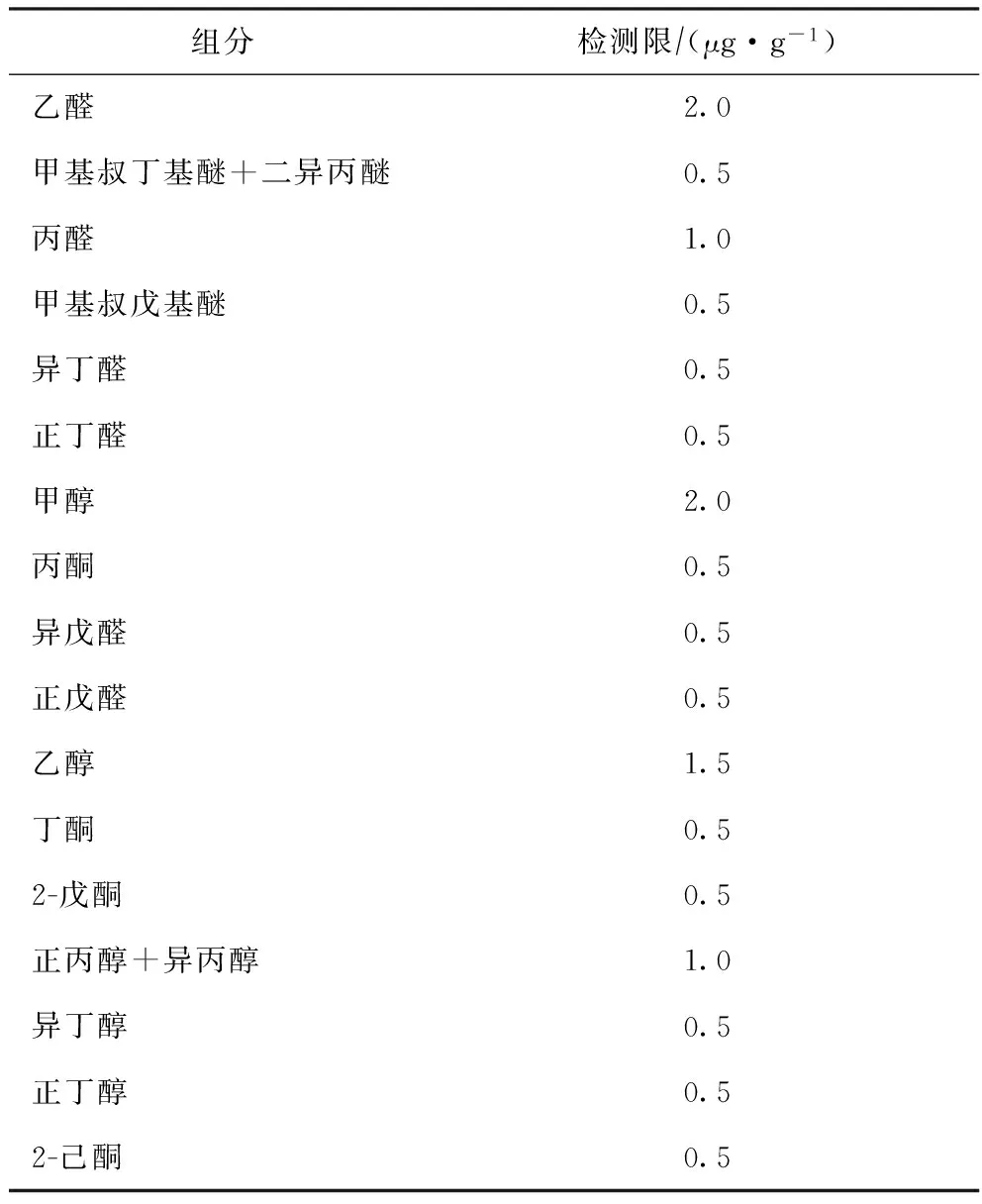
表4 各组分的检测限
2.6 催化裂化及催化裂解等工艺的汽油产品中含氧化合物分布
选择常规催化裂化(FCC)、双提升管催化裂化(FDFCC)、多产异构烷烃的催化裂化(MIP)以及多产丙烯和低碳烯烃的催化裂解(CPP和DCC)工艺的汽油产品,在选定的条件下进行含氧化合物测定,结果见表5。由表5可知:①各工艺的汽油样品中小分子含氧化合物以酮类化合物为主,包括直链酮和异构酮,同时含有少量的醛、醇、醚类组分;②FCC和MIP汽油中含氧化合物的分布较为接近,小于C6的小分子含氧化合物的总量在200 μg/g左右,与CPP和DCC汽油明显不同,CPP汽油中含氧化合物含量最高,分布也最为复杂,小于C6的小分子含氧化合物的总量达3 397.7 μg/g。另外,各工艺的汽油样品中均未检测到明显的甲醇色谱峰,甲醇是对丙烯产品有重要影响的组分,由于目前分析的样品数量有限,在何种条件下会产生甲醇,还需进一步研究;从色谱图中还可以看到一些未知组分的谱峰,需在今后的工作中进一步加以研究确定。

表5 催化裂化和催化裂解工艺的汽油产品中小分子含氧化合物分布 μg/g
3 结 论
(1) 建立了采用带有反吹组件和中心切割组件及3根色谱柱的色谱系统,可以用于测定汽油中微量小分子含氧化合物的含量,通过载气压力切换的方式也可用于直接测定微反产物汽油馏分中微量小分子含氧化合物的含量,样品中所含的大量烃类组分不干扰测定结果。所建立的方法可以测定C1~C4醇、C2~C5醛、C3~C6酮、甲基叔丁基醚、乙基叔丁基醚、甲基叔戊基醚的含量,单组分的检测限为0.5~2.0 μg/g,各组分测定的加标回收率基本在80%~120%之间,测定结果的相对标准偏差在2%~5%的范围内。
(2) 对催化裂化和催化裂解工艺的汽油产品中含氧化合物的分析结果显示,汽油中含小分子含氧化合物以酮类化合物为主,包括直链酮和异构酮,同时含有少量的醛、醇、醚类组分;催化裂解汽油的含氧化合物含量较高,分布较为复杂,小于C6的小分子含氧化合物的总量可达几千μg/g;各工艺的汽油样品中均未检测到明显的甲醇色谱峰。
[1] SH/T 0663.汽油中某些醇类和醚类测定法 气相色谱法[S]
[2] SH/T 0720.汽油中含氧化合物测定法 气相色谱/氧选择性检测器法[S]
[3] McCurry J D,Quimby B D.Two-dimensional gas chromatography analysis of components in fuel and fuel additives using a simplified heart-cutting GC system[J].J Chromatogr Sci,2003,41(10):524-527
[4] Seeley J V,Primeau N J,Bandurski S V,et al.Microfluidic deans switch for comprehensive two-dimensional gas chromatography[J].Anal Chem,2007,79(5):1840-1847
[5] 李长秀,杨海鹰.采用中心切割气相色谱法测定乙苯中微量二甲苯的含量[J].石油化工,2009,38(2):204-208
[6] 李薇,李继文,彭振磊,等.采用微版流路控制技术分析苯乙烯中微量苯[J].石油化工,2010,39(3):331-335
[7] 黄山梅,黄河柳.气相色谱法测定石脑油中含氧化合物含量[J].广东化工,2007,34(9):103-105
DETERMINATION OF TRACE LEVEL LIGHT OXYGENATES IN GASOLINE BY GAS CHROMATOGRAPHY
Li Changxiu,Wang Yamin,Jin Ke
(SINOPECResearchInstituteofPetroleumProcessing,Beijing100083)
A gas chromatographic system equipped with back-flush and heart-cutting (microfluidic deans switch) parts and three connected columns is established for determination of trace level light oxygenates in gasoline products. The oxygenate in gasoline comprising heavy hydrocarbons can directly be analyzed by pressure changes of carrier gas,and the hydrocarbon components in the sample do not interfere with the determination results. Through the pressure changes of carrier gas,the components with the boiling points lower thann-undecane can be eluted from a 2 m long pre-column into a 30 m long non-polar column,meanwhile the heavy fractions are back-flushed to the vent port to leave the system. The components coming into the 30 m long column are separated by switching solenoid valve of heart-cutting parts,only the separated components with boiling point lower than 2-hexanone can be eluted from the non-polar column and enter into a 10 m long polar OxyPLOT column to separate the light oxygenates and hydrocarbons,and then the effluents are detected by flame ionization detector and quantitatively determined with external standard method. Light oxygenate contents such as C1—C4alcohols,C2—C5aldehydes,C3—C6ketones,methyl tert-butyl ether,ethyl tert-butyl ether and methyl tert-amyl ether in gasoline can be analyzed. The detection limit for single component is between 0.5—2.0 μg/g,the adding standard recovery rate for different components is 80%—120%,and the RSD (relative standard deviation) is in the range of 2%—5%. The results of analysis of products from catalytic cracking and catalytic pyrolysis process show that the main oxygenates in these products are ketones with little amount of alcohols,aldehydes,and ethers.
gasoline;oxygenates;heart-cutting;gas chromatography
2016-01-25;修改稿收到日期:2016-03-15。
李长秀,硕士,教授级高级工程师,主要从事气相色谱在石油化工领域的应用研究工作。
李长秀,E-mail:licx.ripp@sinopec.com。
猜你喜欢
中学化学(2022年5期)2022-06-17
时代英语·高一(2019年5期)2019-09-03
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
中国医学装备(2016年6期)2016-12-01
电测与仪表(2016年11期)2016-04-11
电源技术(2015年5期)2015-08-22
河南医学研究(2014年7期)2014-02-27
河南科技(2014年4期)2014-02-27
卫生职业教育(2014年8期)2014-02-16
