二乙酰大黄酸固体分散体的制备以及体外溶出度的测定
2016-04-06 01:24黄家宇母艳华贵州医科大学药学院贵州贵阳550004
中成药 2016年3期
郝 阳, 黄家宇, 张 敏, 李 莉, 白 璐, 母艳华(贵州医科大学药学院,贵州贵阳550004)
二乙酰大黄酸固体分散体的制备以及体外溶出度的测定
郝 阳, 黄家宇, 张 敏, 李 莉*, 白 璐, 母艳华
(贵州医科大学药学院,贵州贵阳550004)
摘要:目的 制备二乙酰大黄酸固体分散体,提高其体外溶出性能。方法 以聚乙烯吡咯烷酮k30(PVP k30)和泊洛沙姆188(Po1oxamer188)为辅料载体,采用研磨法制备二乙酰大黄酸固体分散体,红外测定法和差示扫描量热法对固体分散体进行结构分析。结果 二乙酰大黄酸∶PVP k30∶Po1oxamer188的最佳比例为2∶1∶1,该条件下制备二乙酰大黄酸固体分散体时,其体外溶出度在30 min达到了90%。差示扫描量热法显示,药物与辅料形成共聚物;红外分析显示,二乙酰大黄酸与两种辅料未形成氢键,结构未发生变化。结论 以PVP k30和Po1oxamer188为载体制备的固体分散体能够显著提高二乙酰大黄酸的体外溶出度。
关键词:二乙酰大黄酸;固体分散体;体外溶出度;聚乙烯吡咯烷酮k30;泊洛沙姆188
二乙酰大黄酸(又名双醋瑞因)是大黄酸通过双乙酰化所得到的产物[1],其作为治疗骨关节炎的新药,具有阻止软骨被破坏、促进软骨合成及修复的作用[2]。由于它在水和醇中的溶解度低,体外溶出效果差,生物利用度不理想,因此需通过制剂技术来提高其溶出度。目前,以固体分散体技术提高难溶性药物的溶出度已经得到了广泛应用[3],药物制成固体分散体后,以分子或无定型状态高度分散于水溶性载体中,从而提高药物的溶出速率与溶出度[4]。本实验以水溶性聚乙烯吡咯烷酮k30(PVP k30)和泊洛沙姆188(Po1oxamer188)为辅料[5-7],制备二乙酰大黄酸固体分散体,并对其溶出度进行测定,为将来相关制
剂产品的生产提供技术依据。
1 仪器与试药
1.1 仪器 T6新世纪紫外分光光度计(北京普析通用仪器责任有限公司);ZIS-6G溶出试验仪(天津市天大天发科技有限公司);水浴锅。
1.2 试药 二乙酰大黄酸(纯度>99%);聚乙烯吡咯烷酮k30、泊洛沙姆188均为分析纯。
2 方法
2.1 固体分散体的制备
2.1.1 二乙酰大黄酸固体分散体的制备 按比例称取二乙酰大黄酸、PVP k30以及Po1oxamer188适量,将PVP k30和Po1oxamer188溶解于无水乙醇中,再倒入二乙酰大黄酸固体粉末,50℃水浴蒸干无水乙醇,-20℃冰箱冷冻2 h,真空干燥器干燥过夜,研磨,过100目筛备用[8]。
2.1.2 药物与辅料物理混合物的制备 取二乙酰大黄酸、泊洛沙姆188和PVP k30适量,按2∶1∶1比例混合,即得。
2.2 二乙酰大黄酸溶出度测定
2.2.1 最大吸收波长的选择 称取二乙酰大黄酸对照品适量,磷酸盐缓冲液(pH=7.0)溶解稀释成4 μg/mL的溶液。另取泊洛沙姆188和PVP k30适量,磷酸盐缓冲液溶解稀释,进行紫外扫描。发现二乙酰大黄酸在258 nm处有最大吸收波长,泊洛沙姆188和PVP k30对其最大吸收没有影响。
2.2.2 标准曲线的制备 精密称取二乙酰大黄酸对照品50.30 mg,置于1 000 m L量瓶中,加磷酸盐缓冲液(pH=7.0)适量,超声溶解,定容至刻度,摇匀,即得对照品贮备液。再分别精密量取贮备液适量,置于25 mL量瓶中,磷酸盐缓冲液稀释定容成1.006、2.013、4.026、6.038、8.051、10.064 μg/mL的标准溶液,以磷酸盐缓冲液为空白,在258 nm处测定吸光度值。以质量浓度为横坐标(χ),吸光度为纵坐标(y)进行线性回归,回归方程为y=0.098 7χ+0.011 4,r=0.999 9。表明二乙酰大黄酸在1.006~10.064 μg/mL范围内线性关系良好。
2.2.3 回收率试验 精密量取溶出度测定条件(pH=7.0的磷酸缓冲液,溶出温度[37±0.5]℃,转速100 r/min)下的溶出样品0.5 mL,共9份,置于25 mL量瓶中,同时分别加入质量浓度为50.32 μg/mL的对照品贮备液1.0、2.0、3.0 mL,定容至25 mL,按照“2.2.2”项下方法测定吸光度值,计算回收率。结果,平均回收率为94.99%,ISD为1.78%。
2.2.4 溶出度的测定 采用《中国药典》2010年版第二部附录XC溶出度测定第一法(篮法)测定[9],量取pH 7.0的磷酸缓冲液(脱气)900 mL,作为溶出介质,以溶出温度(37±0.5)℃,转速100 r/min为溶出条件进行测定。再精密称取含二乙酰大黄酸50 mg的固体分散体适量,置于溶出杯中,开始计时,分别于1、2、4、8、20、30 min取样5 mL,0.45 μm微孔滤膜过滤。同时,向溶出杯中补充等量等温的溶出介质,取续滤液稀释10倍,按照“2.2.2”项下方法进行测定,计算药物累积释放百分率,绘制溶出曲线。
2.3 固体分散体物相鉴别
2.3.1 差热分析(DSC) 工作条件为升温范围20~600℃;升温速率10 k/min;参比物为空铝坩埚;气体为氮气;差热量程-1.0~2.5 mW。分别对二乙酰大黄酸原料药、PVP k30、泊洛沙姆188、物理混合物和所制备的固体分散体样品进行分析。
2.3.2 红外光谱分析(II) 波数范围为4 000~400 cm-1,溴化钾压片。将二乙酰大黄酸、PVPk30以及Po1oxamer188所制备的固体分散体与物理混合物进行红外光谱分析。
3 结果
3.1 体外溶出结果及溶出曲线
3.1.1 不同辅料比例下溶出结果 分别将二乙酰大黄酸、PVP k30和泊洛沙姆188按照4∶1∶1、2∶1∶1、2∶1.5∶1.5、1∶1∶1的比例制备固体分散体,测定不同比例下的药物溶出度,并制作溶出曲线,见图1。由图可知,比例为2∶1∶1的固体分散体有较高的溶出速率和载药量,达到预期的实验设计目的,故选定其作为制备固体分散体的最佳辅料比例。

图1 不同比例辅料制备固体分散体的溶出曲线(n=5)
3.1.2 不同挥干温度下溶出结果 将二乙酰大黄酸、PVP k30和泊洛沙姆188按照2∶1∶1的比例制成固体分散体,选择30、40、50℃的水浴温度挥干无水乙醇,制备不同温度下的药物溶出曲线,见图2。由图可知,不同温度条件下挥干乙醇对药物固体分散体的溶出度无明显差异。

图2 不同挥干温度制备固体分散体的溶出曲线(n=5)
3.2 固体分散体的鉴别
3.2.1 差示扫描量热分析 DSC结果表明,二乙酰大黄酸原料药在252.3℃处有一吸热峰;PVP k30在88.1和425.5℃处各有一吸热峰;泊洛沙姆188在57.9和389.7℃处各有一放热峰;药物与载体比为2∶1∶1的固体分散体中,二乙酰大黄酸的吸热峰远远小于原料药,物理混合物中二乙酰大黄酸的吸热峰略有降低,但高于固体分散体,在335℃处有一较强放热峰,说明药物与载体形成低共熔物[10-11],见图3。3.2.2 红外光谱分析 二乙酰大黄酸中含有C=O、COOH 及CH3COO,其中CH3COO中的C=O在1 770 cm-1处有一强吸收峰;COOH中的C=O在1 695 cm-1处有一强吸收峰;C=O在1 680 cm-1处有一强吸收峰。另外,在1 215 cm-1处还有CH3COO中的C-O强吸收峰;PVP k30分子中含有C=O,在1 654 cm-1处有一强吸收峰;Po1oxamer188主要是由丙二醇和环氧丙烷形成的共聚物,在红外2 889和1 112 cm-1处有相对较强的吸收峰,为其亚甲基和醚键的特征峰。对原料药、物理混合物和固体分散体的红外图谱进行比较分析,发现三者在1 695、1 680 cm-1处的强吸收峰都没有发生位移,峰位均无明显变化,推测其与载体未发生化学变化,见图4。
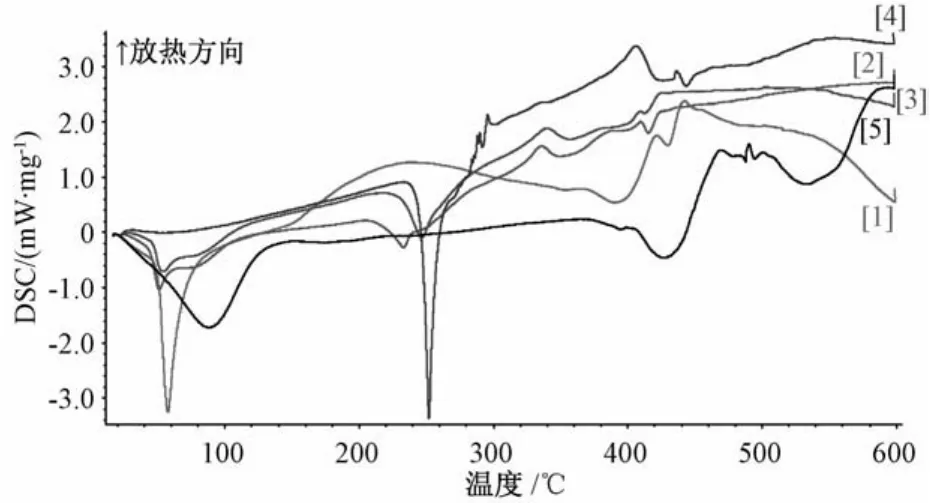
图3 泊洛沙姆188(1)、固体分散体(2)、物理混合物(3)、原料药(4)、PVPk30(5)的差示热分析扫描图
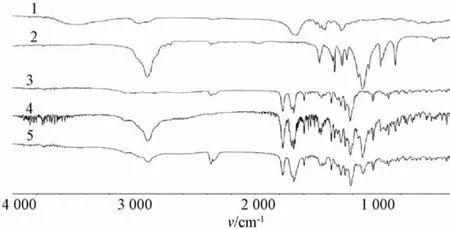
图4 PVPk30(1)、泊洛沙姆188(2)、原料药(3)、物理混合物(4)、固体分散体(5)的红外光谱图
4 讨论
由图1可知,不同比例辅料所制备的固体分散体根据辅料用量的不同,溶出度也有相应变化。当药物、PVP k30、泊洛沙姆188的用量比例为4∶1∶1时,溶出效果较原料药有所增加,但是并未达到最大;当三者用量分别为2∶1∶1、2∶1.5∶1.5、1∶1∶1时,溶出效果显著,而且其体外溶出度无明显差异。为节约成本,本实验选择2∶1∶1作为二乙酰大黄酸固体分散体的制备工艺。由图2可知,不同温度对药物的溶出度没有显著影响,为提高挥干速率,故选择50℃为挥干乙醇的温度。
由于药物熔点较高,高温下稳定性较差,同时二乙酰大黄酸的溶解性也不理想,无法在水或醇中溶解,所以采用研磨法制备固体分散体。同时,以泊洛沙姆188以及PVPk30为固体分散体的载体辅料,可提高原料药的溶出度。由于PVPk30能够以无定形状态存在,而泊洛沙姆188的熔点在52~57℃之间,亲水性强,载药量大,本身也是一种表面活性剂,因此均能很好地应用于固体分散体的制备中。实验显示,选用这两种辅料制备二乙酰大黄酸固体分散体能够显著提高其溶出度。
另外,本品对光线不稳定。实验发现,在药物溶出度达到最大值后,其最大溶出百分率没有在最高点维持,而会略微降低。由此表明,在生产和分析的过程中,应注意避光操作。
参考文献:
[1] 张 毅,王建塔,许 洁,等.一种二乙酰大黄酸的制备方法:中国,201410476405.2[P].2015-01-21.
[2] 黄云台,王 露,杜明瑞,等.双醋瑞因胶囊治疗骨性关节炎的临床研究[J].中外医疗,2010,29(12):82-83.
[3] 张 聪,明 亮.固体分散体技术提高难溶性药物溶出度的研究新进展[J].中国医药指南,2013,11(5):60-61.
[4] 刘娱姗,柯 学.难溶性药物固体分散体研究新进展[J].药学进展,2013,37(4):166-173.
[5] 曾晓丹,刘英姿.大黄酸-泊洛沙姆188固体分散体的制备及释药性能研究[J].湖南师范大学学报:医学版,2013,10(4):92-94.
[6] 韩 刚,王传胜,索 炜,等.大黄酸固体分散体的制备[J].中国医院药学杂志,2011,31(13):1090-1093.
[7] 刘久青,李茂星.大黄游离蒽醌固体分散体的制备及溶出研究[J].中国医药指南,2010,8(4):35-37.
[8] 西里尔·埃斯塔诺夫,阿兰·普吕多姆.以大黄酸或二乙酰大黄酸为基础具有更高生物利用度的药物组合物:中国,97192531.3[P].1999-03-24.
[9] 国家药典委员会.中华人民共和国药典:2010年版二部[S].北京:中国医药科技出版社,2010:附录85-86.
[10] 崔福德.药剂学[M].北京:人民卫生出版社,2007:8.
[11] 张 煤,李章万.依布硒固体分散体的制备及体外溶出度研究[J].华西药学杂志,2005,20(60):512-513.
*通信作者:李 莉(1973—),女,副教授,硕士生导师,研究方向为药物质量控制。Te1:13765110871,E-mai1:1226072965@qq.com
作者简介:郝 阳(1991—),女,硕士,研究方向为药物分析。Te1:18285060152,E-mai1:443811043@qq.com
基金项目:贵阳市科技计划项目([2013204]4-4);2014年贵州省教育厅大学生创新创业(培育)项目(201410660029)
收稿日期:2015-05-15
doi:10.3969/j.issn.1001-1528.2016.03.051
中图分类号:I 944
文献标志码:B
文章编号:1001-1528(2016)03-0702-03
网络出版日期:2015-08-28
网络出版地址:http://www.cnki.net/kcms/detai1/31.1368.I.20150828.1155.004.htm1