
NFATc1 对裸鼠上皮性卵巢癌移植瘤脉管生成的影响*
2016-03-05 02:20段赵宁蔡海贝唐良萏
中国病理生理杂志 2016年2期
龙 丽, 段赵宁, 蔡海贝, 唐良萏
(重庆医科大学附属第一医院,重庆 400016)
·论著·
NFATc1 对裸鼠上皮性卵巢癌移植瘤脉管生成的影响*
龙丽,段赵宁,蔡海贝,唐良萏△
(重庆医科大学附属第一医院,重庆 400016)
[摘要]目的: 探讨胞浆活化T细胞核因子1(nuclear factor of activated T-cells, cytplasmic 1, NFATc1)对人卵巢癌SKOV3细胞裸鼠移植瘤生长和肿瘤脉管生成的影响及其可能机制。方法:NFATc1 siRNA转染人上皮性卵巢癌细胞株SKOV3,免疫荧光及RT-PCR测量转染效率和基因抑制率,选取效率最高的序列建立裸鼠皮下移植瘤模型, 测量各组裸鼠肿瘤体积,观察NFATc1 siRNA的体内抗肿瘤作用。免疫组织化学检测各组肿瘤组织NFATc1的表达情况,并使用细胞角蛋白染色标记上皮性来源,CD34标记微血管,podoplanin标记微淋巴管。分别计算各组微血管及微淋巴管密度并进行统计学分析。应用RT-PCR及Western blot检测各组移植瘤组织NFATc1、CXC趋化因子受体2(CXCR2)、成纤维细胞生长因子2(FGF-2)及血小板源性生长因子BB(PDGF-BB)的mRNA及蛋白表达水平。结果: 3条特异性序列均可显著降低NFATc1的表达水平,以siRNA-1169最佳。NFATc1在空白组及阴性对照组瘤组织高表达。干扰组抑瘤率为57.08%,且重量和体积均低于2个对照组。空白组和阴性对照组的微血管密度和微淋巴管密度明显高于干扰组。对照组比较,NFATc1 siRNA可以在mRNA水平上明显抑制NFATC1、CXCR2、FGF-2和PDGF-BB的转录。Western blot各组细胞在相应位置出现NFATc1、CXCR2、FGF-2和 PDGF-BB条带,空白组与阴性对照组的吸光度最强,与干扰组比较具有显著差异。结论:NFATc1 siRNA明显抑制人卵巢癌SKOV3细胞裸鼠皮下移植瘤生长和肿瘤脉管生成,下调CXCR2、FGF-2及PDGF-BB的表达可能为其途径之一。
[关键词]NFATc1; 上皮性卵巢癌; 肿瘤脉管生成
胞浆活化T细胞核因子1(nuclear factor of activated T-cells, cytoplasmic 1,NFATc1)属于活化T细胞核因子(nuclear factor of activated T-cells,NFAT)家族成员,其功能包括诱导淋巴细胞的发生和激活,以及心肌细胞的分化[1-2]。淋巴细胞的NFAT被激活时,可以单一或异形剪接体的形式与DNA结合,其结合部位可对其他转录因子产生很大的吸引力[3]。例如激活蛋白1(activator protein-1,AP-1)与NFAT和DNA形成复合体,是NFAT激发T细胞活性必需的初始转录元件[4-5]。此外,NFAT和其他许多转录因子一起执行细胞的活动与分化功能,如转录因子GATA-4、早期生长反应蛋白(early growth response protein,EGR)、肌细胞增强因子2(myocyte enhancer factor 2,MEF2)以及在恶性肿瘤中特别重要的叉头盒P1(forkhead box P1,FoxP1)[6]。
最近许多关键的研究发现NFAT与恶性肿瘤的发生发展密切相关[7],目前已明确NFATc1具有致瘤的活性[8]。NFATc1诱导纤维母细胞NIH 3T3产生非常明显的转换表型,被认为是潜在的癌基因[9]。NFATc1的表达与其它许多恶性肿瘤相关,例如在结肠和胰腺癌[10-12],NFATc1是它们发生发展必不可少的关键因子。
NFATc1是胚胎形成的过程中心血管发育的关键因素[13-14],产后它仍可调节内皮细胞的生长,分化和细胞周期。越来越多的数据和资料显示NFATc1调节血管源性的反应[15-16]。对血管内皮生长因子(vascular endothelial growth factor,VEGF)诱导血管生成的研究发现,NFATc1是其关键成分并处于VEGF产生的血管源性内皮细胞逃逸途径的交合点,在VEGF/IL-1诱导大多数基因的过程中非常重要。NFATc1信号途径上游的VEGF-A是肿瘤血管生成的关键因子,同时也具有促进肿瘤淋巴管生成的作用[17]。 此外,NFATc1 在内皮细胞与EGR-1共同激活组织因子(tissue factor,TF)基因转录[18],TF是血液凝固和血管生成的启动蛋白,在TF启动子,NFATc1与NF-κB密切结合于重合位点。这些数据表明VEGF-A对TF及其他潜在基因的完全性转录反应需要NFATc1。NFAHc1在淋巴管生成也具有重要地位,研究发现其表达于发育中及成熟的淋巴管,在淋巴内皮细胞控制podoplanin(肾小球足突细胞膜黏蛋白)和血管内皮生长因子受体3(vascular endothelial growth factor receptor-3,VEGFR-3)的表达,降低其活性可减少肺损伤时VEGF-A所诱导的淋巴管生成[19]。研究还发现NFATc1可作为 VEGF-C 的下游信号分子,与Prox1(prospero-related homeobox 1)、podoplanin、Foxc2 和 VEGFR-3 等促淋巴管生成因子相互作用,影响淋巴管生成,特别是淋巴管的空间构造和管道塑形[20]。以上研究均提示NFATc1在血管及淋巴管生成中有非常重要的地位和作用,但目前NFATc1在恶性肿瘤脉管生成的研究主要集中于其上游基因VEGF,并且在上皮性卵巢癌还没有相关的研究。因此,在本研究中我们从体内外两个方面探讨NFATc1对上皮性卵巢癌血管及淋巴管生成的影响,以及NFATc1与对肿瘤血管及淋巴管生成具有非常重要作用的CXC趋化因子受体2(CXC chemokine receptor 2,CXCR2)、成纤维细胞生长因子2(fibroblast growth factor 2,FGF-2)和血小板源性生长因子BB(platelet-derived growth factor-BB,PDGF-BB)的关系,以期解释NFATc1影响上皮性卵巢癌恶性生物学行为的可能原因,探索NFATc1的信号转导和上皮性卵巢癌发生发展的分子机制,为肿瘤脉管生成提供更多的理论基础。
材料和方法
1实验材料与分组
1.1细胞人上皮性卵巢癌细胞株SKOV3(人卵巢浆液性乳头状囊腺癌系)由重庆医科大学分子医学与肿瘤中心惠赠。设置为5个组:空白组不予干扰;阴性对照组给予无关序列;干扰组1(siRNA1)给予siRNA-880;干扰组2(siRNA2)给予siRNA-1169;干扰组3(siRNA3)给予siRNA-1307。
1.2实验动物4~8周龄雌性裸鼠(n=18)由重庆医科大学动物实验中心提供,平均设置为3个组。空白组种植未加干扰的SKOV3细胞;阴性对照组种植无关序列干扰的SKOV3细胞;干扰组种植抑制效率较高的NFATc1 siRNA 干扰后的SKOV3细胞。
1.3主要抗体和工作浓度NFATc1兔多克隆抗体(Abcam)的工作浓度为1∶500;CXCR2兔抗人多克隆抗体(Santa Cruz)为1∶800;FGF-2鼠单克隆抗体 (Santa Cruz)为1∶500;PDGF-BB兔多克隆抗体(Santa Cruz)为1∶800。
2实验方法
2.1细胞培养与转染人卵巢癌SKOV3细胞株置于RPMI-1640培养基。取对数生长期细胞,按每孔1×105接种于 6孔板,长至70%~80%融合时进行转染:250 μL的无抗无血清DMEM稀释5 μL的脂质体,室温放置5 min;250 μL的无抗无血清DMEM稀释5 μL的浓度为20 μmol/L的siRNA。将稀释后的脂质体和siRNA混在一起,共510 μL,室温放置20 min。PBS洗细胞3次,每孔加入1.5 mL无抗培养基,然后再加入510 μL 的siRNA/脂质体复合体混匀,放入培养箱培养24 h后荧光显微镜及RT-PCR观察转染情况。 经3条特异性干扰序列处理的细胞在高倍镜下各自随机选取5个视野,转染效率按以下公式计算:转染效率(%)=发绿色荧光细胞平均数/总细胞平均数×100%。RT-PCR半定量测定基因抑制情况。
2.2动物模型的建立4~6周龄雌性裸鼠饲养在 SPF环境中。收集处于对数生长期的各组SKOV3细胞,于每只裸鼠侧腋皮下注射单细胞悬液(5×106个,0.2 mL)。连续观察30 d,记录肿瘤出现的时间,定期观察小鼠精神、饮食、排便及成瘤情况,于接种后第30 d断颈处死各组裸鼠,摘取移植瘤称取重量,以瘤体重量计算抑瘤率:抑瘤率(%)=(对照组重量-实验组重量)/对照组重量×100%;计算肿瘤体积(V):V (cm3)=1/2 × LW2(L:长度;W:重量)。RT-PCR和Western blot实验观察各组移植瘤瘤体NFATc1 mRNA及蛋白表达情况。
2.3免疫组织化学观察石蜡包埋组织6 μm连续切片,每例标本连续切片3张。采用免疫组化链霉菌抗生物素蛋白-过氧化物酶连结法(SP法)分别检测每组标本血管标记物CD34及淋巴管标记物podoplanin的表达。每批切片均设阳性和阴性对照片,阴性对照采用 PBS替代第I抗体, 已知阳性片为阳性对照。使用的抗体即广谱细胞角蛋白(cytokeratin,CK)鼠单克隆抗体,工作浓度为1∶500;CD34兔多克隆抗体,工作浓度为1∶500;podoplanin鼠单克隆抗体,工作浓度为1∶1 000。
以CD34为血管内皮标志物,以podoplanin为淋巴管内皮标志物,并在光镜下观察着色血管内皮细胞与着色淋巴管内皮细胞的数量,微血管微淋巴管形态。每例先在光镜低倍视野(×40)下观察,选取着色细胞密集的区域,在高倍视野(×200或×400)下观察,鉴别着色的淋巴管并计数。以5个200倍或400倍视野内淋巴管数的平均值与镜下面积相除得出该例切片的微淋巴管密度。每张切片均由2位病理科医生双盲法阅片。
2.4RT-PCR实验充分裂解细胞后提取RNA,逆转录后按下列程序分别扩增NFATc1、CXCR2、FGF-2及PDGF-BB:95 ℃ 5 min;56 ℃(NFATC1)或57 ℃(CXCR2)或58 ℃(FGF-2和PDGF-BB)。并按下列程序扩增内参照GAPDH:95 ℃,5 min;59.5 ℃,30 s,30 cycles;72 ℃ 30 s;72 ℃ 10 min。反应结束后,取5 μL扩增产物作1.5%琼脂糖凝胶电泳,用Quantity One凝胶成像系统分析各基因灰度值。RT-PCR的引物序列见表1。
2.5.Western blot实验转染48 h后收集细胞。充分裂解细胞20 min;4 ℃,15 000 r/min离心15 min,吸取上清并用6(buffer稀释,99 ℃高温下变性5 min,用6×加样缓冲液1∶5稀释蛋白样品,每孔30 μL上样,72 V 30 min,然后2 h行SDS-PAGE至溴酚蓝达到凝胶底部。根据蛋白大小切割胶及PVDF(Millipore)膜,冷却条件下250 mV电转60 min;封闭液稀释 I 抗NFATc1/CXCR2/PDGF-BB兔多克隆抗体(1∶1 000)和FGF-2鼠单克隆抗体(1∶1 000),4 ℃过夜;加入稀释好的HRP(浓度为1∶2 000)标记的山羊抗兔 II抗显色。

表1 RT-PCR的引物序列
F: forward; R: reverse.
3统计学处理
所有的统计学处理均采用 SPSS 16.0软件完成。计量资料用均数(标准差(mean±SD)表示。多样本均数比较首先采用单因素方差分析(one-way ANOVA),多样本各组均数间的多重比较采用SNK-q检验。以P<0.05为差异具有统计学意义。
结果
1转染效率
转染后24 h及48 h荧光显微镜观察,可见SKOV3细胞内出现绿色荧光颗粒,说明siRNA已经被转染进入细胞(图1),各组48 h转染效率均高于24 h,siRNA1组48 h转染效率为59.1%,siRNA2组转染效率为85.3%,siRNA3组转染效率为51.9%。以siRNA2组48 h转染效率最高,与其余2组比较差异有统计学显著性。
RT-PCR 检测3组siRNA 转染48 h对NFATc1基因的抑制情况,结果显示3组细胞NFATc1 mRNA表达水平分别为0.532±0.001、0.278±0.001和0.498±0.003,siRNA2组siRNA抑制效率最高,与其余2组比较差异均有统计学显著性(P<0.01),siRNA1组与siRNA3组间比较无显著差异(图2)。根据对3条siRNA转染及抑制效率的比较,在后续试验中采用效率最高的siRNA-1169进行干扰。
Figure 1.Transfection efficiency of each group observed under fluorescent microscope. Mean±SD.n=24.*P<0.05vssiRNA1 or siRNA3.
图1荧光显微镜分别观察各组转染效率

Figure 2.Inhibitory efficiency of each siRNA at 48 h. Mean±SD.n=9.**P<0.01vssiRNA1 or siRNA3.
图2各组siRNA 转染48 h后NFATc1的mRNA表达水平
2降低NFATc1的活性可抑制裸鼠移植瘤生长
18只裸鼠接种肿瘤成功率为100%,空白对照组和阴性对照组潜伏期6~8 d,NFATc1 siRNA组潜伏期8~11 d。肿瘤在局部呈膨胀性生长,2~3周后体积逐渐增大(图3)。实验结束时裸鼠均成活。肉眼观察NFATc1 siRNA组肿瘤体积较对照组小。

Figure 3.The nude mouse model of transplanted ovarian cancer. A: nude mouse; B: transplanted tumorinvivo; C: isolated transplanted tumor.
图3人卵巢癌裸鼠移植瘤模型
NFATc1 siRNA组抑瘤率为57.08%,且重量和体积均小于2个对照组,差异有统计学显著性,见表2。
表2各组裸鼠移植瘤瘤体平均质量分析
Table 2.Analysis of the transplanted tumor of nude mouse (Mean±SD.n=18)

GroupWeight(g)Volume(cm3)Inhibitoryrate(%)Blankcontrol2.03±0.35*1.328±145*0*Negativecontrol1.98±0.78*1.274±209*0*siRNA0.87±0.320.512±08757.08
*P<0.05vssiRNA.
3NFATc1在移植瘤组织高表达
每组切片30张,共90张切片进行染色,NFATc1的抗体效应物为棕黄色颗粒,定位于细胞质。阳性表达于全部对照组移植瘤组织(100%),H-Score半定量法平均积分为320(0~356)和336(0~356);阳性表达于10例(33%)干扰组移植瘤组织,H-Score平均积分为35(0~180)。NFATc1在上皮性卵巢癌移植瘤组织高表达,NFATc1 siRNA可明显抑制其表达水平,与对照组比较差异有统计学显著性(P<0.05),见图4、表3。
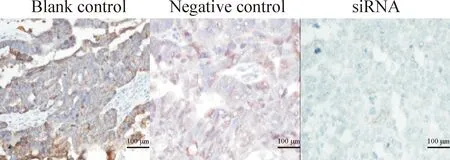
Figure 4.The protein expression of NFATc1 in the mouse transplanted tumors.
图4NFATc1蛋白在裸鼠移植瘤组织的表达
4降低NFATc1的活性可抑制上皮性卵巢癌裸鼠移植瘤组织血管及淋巴管生成
CK呈棕黄色特异性表达于所有移植瘤组织切片上皮细胞的胞浆,证明了移植瘤的上皮性来源。CD34特异性地表达于移植瘤组织毛细血管内皮细胞胞质,标记毛细血管为棕黄色管腔,管腔内见或不见红细胞。肿瘤细胞未见表达。Podoplanin染色后微淋巴管内皮细胞胞质呈棕黄色,标记微淋巴管为棕黄色管腔。淋巴管管壁较血管壁薄,腔相对较大而塌陷,腔内不见红细胞。染成棕黄色的单个内皮细胞或内皮细胞簇连成闭合或不闭合的线状管腔作为一个脉管,以计算微血管密度(microvessel density,MVD)或微淋巴管密度(lymphatic microvessel density,LMVD)。统计结果显示对照组密度明显多于干扰组,差异有统计学显著性,见图5、表3。
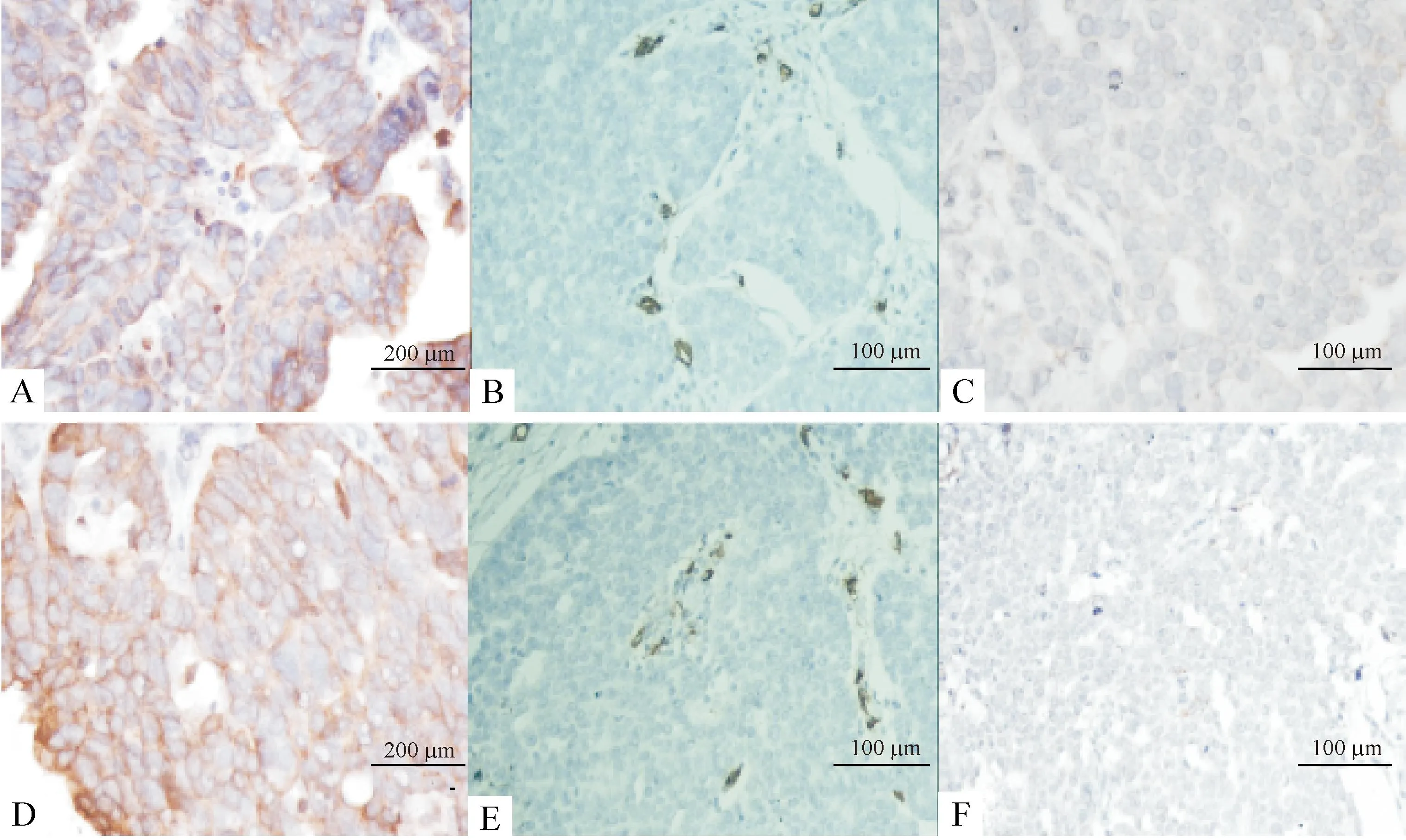
Figure 5.The MVD of transplanted tumor. A, D: epithelial specific CK staining (+++); B: CD34 staining in control group; C: CD34 staining in siRNA group; E: podoplanin staining in control group; F: podoplanin staining in siRNA group.
图5各组上皮性卵巢癌裸鼠移植瘤微血管及微淋巴管密度

表3 各组移植瘤免疫组织化学染色结果、H-Score、MVD和LMVD的比较
H-Score=ΣPi(i+1):iwas the intensity of staining, Piwas the percentage of cells with positive staining/the total number of tested cells, and 1 was the correction factor.*P<0.05vssiRNA.
5沉默NFATc1可抑制SKOV3细胞CXCR2、FGF-2和PDGF-BB的mRNA表达
扩增各组NFATc1、CXCR2、FGF-2和PDGF-BB电泳条带存在明显差异。其中,空白组和阴性对照组扩增带亮度较强;NFATc1 siRNA组扩增带亮度较弱,用Quantity One凝胶成像系统分析NFATc1、CXCR2、FGF-2、PDGF-BB与内参照β-actin的灰度比值,与2个对照组比较,差异均有统计学显著性(P<0.05),见表4。这一结果证实NFATc1 siRNA可以在mRNA水平上明显抑制上述4种基因的转录。
6沉默NFATc1可抑制SKOV3细胞CXCR2、FGF-2和PDGF-BB蛋白的表达
各组细胞在相应位置均出现分别为91 kD的NFATc1、40 kD的CXCR2、15.8 kD的FGF-2和24.4 kD的 PDGF-BB条带,但强弱不一,空白组与阴性对照组表达最强,干扰组表达最弱,各组与干扰组比较差异有统计学显著性(P<0.05),见图6。

表4 各组SKOV3细胞NFATc1、CXCR2、FGF-2和PDGF-BB mRNA的表达
*P<0.05vssiRNA.

Figure 6.The protein expression of NFATc1, CXCR2, FGF-2 and PDGF-BB in each group. Mean±SD.n=24.*P<0.05vssiRNA.
图6各组NFATc1、CXCR2、FGF-2及PDGF-BB蛋白表达的比较
讨论
有报道NFATc1诱导肺动脉内皮细胞增殖[21],并发现其增加人肺动脉瓣内皮细胞的迁移和移行[22]。NFATc1还是胚胎形成过程中心血管发育的关键因素,通过与特异性的配体辅助因子(如MEK1-ERK1/2 和JNK1/2)协作促进其有序发育[23]。VEGF/NFATc1/COX2信号途径也可显示NFATc1在血管及淋巴管生成中的重要地位[24-25]。
通过建立人卵巢癌裸鼠移植瘤模型取得移植瘤组织,并采用CD34染色肿瘤微血管,podoplanin染色肿瘤微淋巴管,免疫组化观察NFATc1的表达及NFATc1 siRNA作用前后移植瘤血管及淋巴管生成,可见NFATc1在移植瘤组织高表达,用 siRNA抑制NFATc1的表达可显著减少上皮性卵巢癌血管生成和淋巴管生成,说明NFATc1可促进上皮性卵巢癌血管生成和淋巴管生成,在上皮性卵巢癌的脉管生成中具有重要作用。
PCR结果显示干扰组NFATc1、CXCR2、FGF-2和PDGF-BB的基因表达水平明显低于对照组。NFATc1 siRNA作用于SKOV3细胞后明显抑制NFATc1基因水平,同时导致CXCR2、FGF-2和PDGF-BB表达下降,Western blot实验结果与PCR实验一致,NFATc1 siRNA作用于SKOV3细胞后明显抑制NFATc1蛋白水平,同时导致CXCR2、FGF-2和PDGF-BB 蛋白表达下降。这些实验结果均提示NFATc1 siRNA不但可以抑制上皮性卵巢癌SKOV3细胞NFATc1 mRNA和蛋白表达,还可以抑制CXCR2、FGF-2和PDGF-BB 的mRNA和蛋白表达水平。
CXCR2也称为IL-8受体, 属于G蛋白偶联跨膜趋化因子受体[26],CXC趋化因子与其结合对血管生成产生影响。CXC趋化因子的区分基于ELR(N 端谷氨酸-亮氨酸-精氨酸基序)的表达,ELR+CXC 趋化因子生成血管,其代表有CXCL8/IL-8,是血管生成强烈的诱导子和主要的基础。ELR+CXC趋化因子在血管生成中的关键性使其成为恶性肿瘤发生发展中血管生成异常调节的基础[27-30]。CXCR2即ELR+CXC 趋化因子的特异性受体,通过与ELR+CXC 趋化因子结合实现其癌组织血管生成的功能,此外,它还可以通过与多种信号途径,如PI3K/Akt、NF-κB、MAPK和STAT3作用实现肿瘤血管生成,为其在恶性肿瘤发生发展中扮演关键角色提供了有力的证据[31]。在本实验中,通过NFATc1 siRNA干扰的手段发现CXCR2的表达水平随着NFATc1的降低而降低,说明NFATc1对上皮性卵巢癌血管生成的影响或许可以通过调节CXCR2来实现。
恶性肿瘤积极的产生淋巴管生成和塑形,肿瘤诱导的淋巴管生成是由淋巴管生成因子发动,其中FGF-2 和PDGF-BB是非常重要的2个因子。既往的研究发现FGF-2是促进血管生成的生长因子,有力的有丝分裂原和化学趋化剂,近年的研究确定其与肿瘤淋巴管生成及淋巴转移密切相关,当其低剂量表达时不能诱导血管生成,但是却能够特异性的诱导肿瘤淋巴管生成,因此在肿瘤淋巴管生成中扮演重要角色[32]。PDGF-BB是PDGFs中的一员,研究显示PDGF-BB可以刺激肿瘤淋巴管生成及肿瘤的淋巴转移,是VEGF-C之外独立的淋巴管生成因子,其主要功能是稳定脉管网络,最近发现它还可以独立于VEGFR-3直接刺激肿瘤淋巴管生成及淋巴转移[33],特异性的靶向PDGF-BB是抑制淋巴管生成新的策略。本实验通过NFATc1 siRNA干扰发现NFATc1可影响FGF-2 和PDGF-BB,两者表达水平随着NFATc1的降低而降低,说明NFATc1对上皮性卵巢癌淋巴管生成的影响或许可以通过调节FGF-2 和PDGF-BB来实现。
此外,COX-2促进血管生成和肿瘤侵袭过程中的激活会增加PDGF和bFGF的表达水平[34],并且VEGF对PDGF的激活依赖于COX2的过表达[35],在内皮细胞,刺激FGF-2可以获得PDGF-BB 的高反应性,反过来PDGF-BB会通过放大淋巴管壁细胞的受体表达对FGF-2信号产生正反馈。二者这种不协调的相互交集在肿瘤微环境中会引起原始淋巴管的无序形成和塑形,从而促进肿瘤生长和转移。结合VEGF/NFATc1/COX2信号途径与我们的研究结果,或许可以认为在上皮性卵巢癌脉管生成的过程中,NFATc1具有非常重要的作用:当其受VEGF-A或VEGF-C的激活后,作用于其下游的IL-8促进血管生成,同时,通过其下游的COX2,影响FGF-2及PDGF-BB的水平,从而调控肿瘤血管和淋巴管生成,且FGF-2和PDGF-BB二者间形成正反馈环,相互作用相互影响,通过肿瘤脉管生成共同促进癌发生及癌转移,该信号通路有可能是上皮性卵巢癌的有效治疗靶标。
[参考文献]
[1]Naito T, Tanaka H, Naoe Y, et al. Transcriptional control of T-cell development[J]. Int Immunol, 2011, 23(11):661-668.
[2]Rinne A, Blatter LA. Activation of NFATc1 is directly mediated by IP3in adult cardiac myocytes[J]. Am J Physiol Heart Circ Physiol, 2010, 299(5):H1701-H1707.
[3]Jain J, McCaffrey PG, Valge-Archer VE, et al. Nuclear factor of activated T cells contains Fos and Jun[J]. Nature, 1992, 356(6372):801-8040.
[4]Bengsch B, Wherry EJ. The importance of cooperation: partnerless NFAT induces T cell exhaustion[J]. Immunity, 2015, 42(2):203-205.
[5]Shaw JP, Utz PJ, Durand DB, et al. Identification of a putative regulator of early T cell activation genes[J]. Science, 1988, 241(4862):202-205.
[6]Bai S, Kerppola TK. Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy [J]. Mol Cell Biol, 2011, 31(14):3068-3080.
[7]Oller J, Alfranca A, Méndez-Barbero N,et al. C/EBPβ and nuclear factor of activated T cells differentially regulate Adamts-1 induction by stimuli associated with vascular remodeling[J]. Mol Cell Biol, 2015, 35(19):3409-3422.
[8]Chen NM, Singh G, Koenig A, et al. NFATc1 links EGFR signaling to induction ofSox9 transcription and acinar-ductal transdifferentiation in the pancreas[J]. Gastroenterology, 2015, 148(5):1024-1034.e9.
[9]Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17[J]. Rheumatology (Oxford), 2008, 47(11):1635-1640.
[10]Wang S, Kang X, Cao S, et al. Calcineurin/NFATc1 pathway contributes to cell proliferation in hepatocellular carcinoma[J]. Dig Dis Sci, 2012, 57(12):3184-3188.
[11]Beals CR, Sheridan CM, Turck CW, et al. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3[J]. Science, 1997, 275 (5308):1930-1934.
[12]Köenig A, Linhart T, Schlengemann K, et al.NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells[J]. Gastroenterology, 2010, 138(3):1189-1199.e2.
[13]Wu B, Baldwin HS, Zhou B. Nfatc1 directs the endocardial progenitor cells to make heart valve primordium[J].Trends Cardiovasc Med,2013,23(8):294-300.
[14]Wu B, Wang Y, Lui W, et al. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation[J]. Circ Res, 2011, 109(2):183-192.
[15]Adinolfi E, Raffaghello L, Giuliani AL, et al. Expression of the P2X7 receptor increasesinvivotumor growth[J]. Cancer Res, 2012, 72(12):2957-1969.
[16]Shoemaker LD, Fuentes LF, Santiago SM, et al. Human brain arteriovenous malformations express lymphatic-associated genes[J]. Ann Clin Transl Neurol, 2014, 1(12):982-995.
[17] Chen HM, Tsai CH, Hung WC. Foretinib inhibits angiogenesis,lymphangiogenesis, lymphangiogenesis and tumor growth of pancreatic cancerinvivoby decreasing VEGFR-2/3 and TIE-2 signaling[J]. Oncotarget, 2015, 6(17):14940-14952.
[18]KähäräJ, Lähdesmäki H. Evaluating a lineark-mer model for protein-DNA interactions using high-throughput SELEX data[J]. BMC Bioinformatics, 2013,14(Suppl 10):S2.
[19]Kulkarni RM,Greenberg JM,Akeson AL. NFATc1 regulates lymphatic endothelial development[J]. Mech Dev, 2009, 126(5-6):350-365.
[20]Norrmén C,Ivanov KI,Cheng J,et al. FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1[J]. J Cell Biol, 2009, 185(3):439-457.
[21]Lee JH, Bhang DH, Beede A, et al. Lung stem cell diffe-rentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis[J]. Cell, 2014, 156(3):440-455.
[22]Jang GH, Park IS, Yang JH,et al. Differential function of genes regulated by VEGF-NFATc1 signaling pathway in migration of pulmonary valve endothelial cells[J]. FEBS Lett, 2010, 584(1):141-146.
[23]Combs MD. Yutzey KE. VEGF and RANKL regulation of NFATc1 in heart valve development[J]. Circ Res, 2009,105(6):565-574.
[24]Suehiro J, Kanki Y, Makihara C,et al. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium[J]. J Biol Chem, 2014, 289(42):29044-29059.
[25]Wang L, Wang Z, Li J, et al. NFATc1 activation promotes the invasion of U251 human glioblastoma multiforme cells through COX-2[J]. Int J Mol Med, 2015, 35(5):1333-1340.
[26]Baird AM, Gray SG, O′Byrne KJ. Epigenetics underpinning the regulation of the CXC (ELR+) chemokines in non-small cell lung cancer[J]. PLoS One, 2011, 6(1):e14593.
[27]Moldobaeva A, Baek A, Eldridge L, et al. Differential activity of pro-angiogenic CXC chemokines[J]. Microvasc Res, 2010, 80(1):18-22.
[28]Joimel U, Gest C, Soria J, et al. Stimulation of angioge-nesis resulting from cooperation between macrophages and MDA-MB-231 breast cancer cells: proposed molecular mechanism and effect of tetrathiomolybdate[J]. BMC Cancer, 2010,10:375.
[29]Sarmiento J, Shumate C, Suetomi K, et al. Diverging mechanisms of activation of chemokine receptors revealed by novel chemokine agonists[J]. PLoS One. 2011, 6(12):e27967.
[30]Rabquer BJ, Tsou PS, Hou Y, et al. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis[J]. Arthritis Res Ther, 2011, 13(1):R18.
[31]Dong YL, Kabir SM, Lee ES, et al. CXCR2-driven ovarian cancer progression involves upregulation of proinflammatory chemokines by potentiating NF-κB activation via EGFR-transactivated Akt signaling[J]. PLoS One,2013, 8(12):e83789.
[32]Chang LK,Garcia-Cardea G, Farnebl F, et al. Dose-dependent response of FGF-2 for lymhangiongenesis [J]. Proc Natl Acad Sci U S A, 2004, 101(32):11658-11663.
[33]Schoppmann SF, Alidzanovic L, Schultheis A, et al. Thrombocytes correlate with lymphangiogenesis in human esophageal cancer and mediate growth of lymphatic endothelial cellsinvitro[J]. PLoS One, 2013, 8(6):e66941.
[34]Tung HC, Lee FY, Wang SS, et al. The beneficial effects of P2X7antagonism in rats with bile duct ligation-induced cirrhosis[J]. PLoS One, 2015, 10(5):e0124654.
[35]Zhao Y, Wang Y, Wang Q, et al. Hepatic stellate cells produce vascular endothelial growth factor via phospho-p44/42 mitogen-activated protein kinase/cyclooxygenase-2 pathway[J]. Mol Cell Biochem, 2012, 359(1-2):217-223.
(责任编辑: 陈妙玲, 罗森)
NFATc1 promotes vascular generation of epithelial ovarian cancer transplanted tumor by regulating CXCR2, FGF-2 and PDGF-BBLONG Li, DUAN Zhao-ning, CAI Hai-bei, TANG Liang-dan
(TheFirstAffiliatedHospitalofChongqingMedicalUniversity,Chongqing400016,China.E-mail:ldtangcq2002@163.com)
[ABSTRACT]AIM: To investigate the role of NFATc1 in vascular generation in the nude mice transplanted with human ovarian cancer SKOV3 cells. METHODS:NFATc1 expression was silenced by siRNA in SKOV3 cells. Human ovarian cancer transplantation nude mouse model was established by transplanting with SKOV3 cells in which theNFATc1 gene was silenced by siRNA technique. The expression of NFATc1, CXCR2, FGF-2 and PDGF-BB at mRNA and protein levels was determined by RT-PCR, Western blotting and immunohistochemical staining. The tumor growth, angiogenesis and lymphangiogenesis were also observed. RESULTS: Over-expression of NFATc1 was observed in human ovarian cancer tissues. The silencing ofNFATc1 expression by siRNA decreased tumorigenesis of transplanted ovarian cancer cells in the nude mice, reduced tumor vascular generation and inhibited the expression of CXCR2, FGF-2 and PDGF-BB at mRNA and protein levels. CONCLUSION: NFATc1 is overexpressed in ovarian cancer.NFATc1 silencing regulates the tumor vascular generation. NFATc1 thus has potential as a therapeutic target and for use in the diagnosis and evaluating prognosis of epithelial ovarian cancer.
[KEY WORDS]NFATc1; Epithelial ovarian cancer; Tumor vascular generation
doi:10.3969/j.issn.1000- 4718.2016.02.001
[中图分类号]R730.23;R711
[文献标志码]A
通讯作者△Tel: 023-89011080; E-mail: ldtangcq2002@163.com
*[基金项目]国家自然科学基金资助项目(No. 81402126)
[收稿日期]2015- 07- 15[修回日期] 2015- 12- 11
[文章编号]1000- 4718(2016)02- 0193- 08
