
原发性皮肤间变性大细胞淋巴瘤1例
2016-03-03 05:39严志陈柳青张良
中国中西医结合皮肤性病学杂志 2016年6期
严志,陈柳青,张良
(湖北省武汉市第一医院,武汉430022)
原发性皮肤间变性大细胞淋巴瘤1例
严志,陈柳青,张良
(湖北省武汉市第一医院,武汉430022)
患者男,59岁。下颌反复出现红斑2年余。皮损病理组织示:角化过度,角化不全,表皮增生,表皮基底部细胞向下增生,细胞排列轻度紊乱,可见异常核分裂相,真皮乳头血管增生扩张,浅深层可见单一核细胞浸润,细胞体积大,核异型性明显。免疫组化染色结果显示:CD3(+)、CD4(+)、CD8(+)、CD20(+)、CD79a(+)、CD30(+)、S-100(-)、CK20(-)、HCK(-)、Ki-67阳性细胞约50%~60%。诊断为原发性皮肤间变性大细胞淋巴瘤。
间变性;大细胞;淋巴瘤
原发性皮肤间变性大细胞淋巴瘤(Primary cutaneous anaplastic cell lymphoma,C-ALCL)临床上较罕见,现将我科诊治的1例报告如下。
1 临床资料
患者男,59岁。因下颌反复出现红斑2年余,于2014年12月22日入住我院。患者20年前不明诱因发现下颌部成簇白色丘疹,自行搔抓后反复破溃,逐渐自愈,多年未复发,约2年余前下颌部不明诱因出现1处境界清晰浸润性红斑,形状不规则,表面散在薄层黄痂,片状糜烂面,较潮湿,红斑边缘隆起增厚,围绕红斑边缘可及较薄表皮,皮损触之较硬,曾于当地医院行抗炎、免疫调节及对症治疗,皮损缩小后又再次增大。发病以来,无明显消瘦、贫血和食欲不振等,饮食及睡眠正常。既往体健,无不良嗜好,其直系亲属无类似症状。一般情况较好,系统查体未及明显异常。皮肤科情况:下颌部1处境界清晰浸润性红斑,形状不规则,约3 cm×5 cm,表面散在薄层黄痂,点片状糜烂面,少许渗出,红斑边缘稍隆起,见图1,时有瘙痒,偶有刺痛。实验室及辅助检查:血、尿、粪常规和肝肾功能均正常;糖类抗原及尿β2微球蛋白升高;双侧颈部、腋窝及腹股沟淋巴结彩超有显示;胸腹部CT显示肝肾钙化结石灶,余正常。皮损组织病理检查:角化过度,角化不全,表皮增生,表皮基底部细胞向下增生,细胞排列轻度紊乱,可见异常核分裂相,真皮乳头血管增生扩张,浅深层可见单一核细胞浸润,细胞体积大,核异型性明显,见图2、3。免疫组化染色结果显示:CD3(+)、CD4(+)、CD8(+)、CD20(+)、CD79a(+)、CD30(+)、S-100(-)、CK20(-)、HCK(-)、Ki-67阳性细胞约50%~60%,结合临床表现诊断为CD30阳性大细胞性淋巴瘤。骨髓穿刺检查结果回报:骨髓淋巴细胞中幼淋占4.5%,但流式细胞学分型未见明显T细胞克隆增生,未见明显侵犯骨髓,故考虑多为原发于皮肤局部的淋巴瘤。入院后完善相关检查后行免疫调节治疗。2014年12月29开始肌肉注射干扰素α-2a 1百万单位,3次/周,以治疗皮损。2015年1月6日第4次肌肉注射干扰素α-2a,剂量增至3百万单位,3次/周。2015年1月9日开始以1百万单位干扰素α-2a皮损局部注射,每周3次。患者于干扰素α-2a肌肉注射治疗3次后皮损即明显消退,干扰素α-2a皮损局部注射3次后,皮损基本消退。随访2个月病情未复发。

图1 原发性皮肤间变性大细胞淋巴瘤患者下颌部皮损
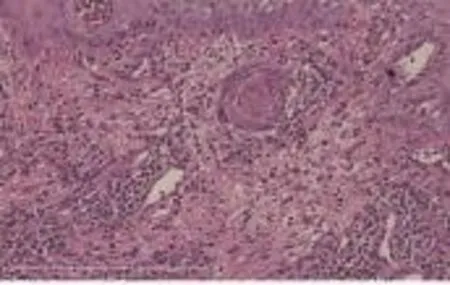
图2 皮损组织病理像(HE染色×200)
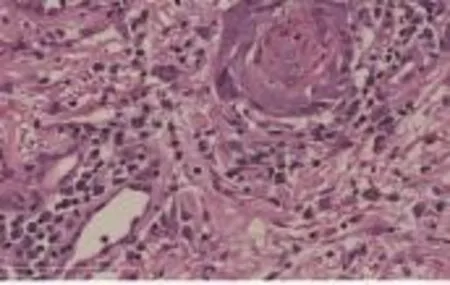
图3 皮损组织病理像(HE染色×400)
2 讨论
C-ALCL属于原发性皮肤CD30(+)淋巴增生性疾病,其特征是大于75%的肿瘤细胞表达CD30抗原,发病机制不明。原发性C-ALCL占所有CTCL的10%,好发于成年人,偶见于儿童或青少年,男女发病率之比为2∶1。皮损表现为单发性或局限性的结节或肿瘤,有时为丘疹,皮损表面发生溃疡,20%的患者表现为多发性损害。皮损可部分或完全消退,但常复发。10%患者有其他脏器受累,首选受累的器官是局部淋巴结。预后通常很好,但长期随访很重要。组织病理学特点为瘤细胞体积大,胞浆丰富,核呈马蹄形或肾形,细胞有间变性或异型性免疫母细胞性,瘤细胞侵犯真皮及皮下组织,无明显亲表皮现象[1]。免疫表型方面,大部分肿瘤细胞特征性表达CD30,CD4常为阳性,CD2、CD5和CD3常缺失。大部分原发性C-ALCL表达皮肤淋巴细胞抗原(CLA),而不表达上皮膜抗原(EMA)或间变性淋巴瘤激酶(ALK),这一点可与系统性CD30(+)淋巴瘤鉴别。C-ALCL大部分患者显示TCR基因重排[2]。
诊断原发性C-ALCL尚应鉴别及除外淋巴瘤样丘疹病(LyP)、继发于蕈样肉芽肿(MF)的CD30(+)大细胞淋巴瘤,以及伴有皮肤受累的系统性ALCL。C-ALCL和LyP都属于原发性皮肤CD30(+)淋巴增生性疾病,两者在临床、病理及免疫表型上有一定的重叠性,形成一谱系,仅根据组织学改变很难区分两者,但两者临床表现和发展过程不同,有利于诊断和治疗。LyP的特征性皮损为出血和坏死性丘疹和结节,4~8周皮损可自然消退,尽管有新发皮损,也应诊断为LyP。累及皮肤的系统性ALCL的诊断要点为:伴发其他脏器的受累;常表现为泛发性皮损;免疫表型为CLA(-)、EMA(+)、ALK(+)。而向大细胞转化的MF的诊断要点为:组织学上伴有典型脑回状细胞浸润;有前驱的或伴发的斑片或斑块的临床表现。
放疗或切除是具有单一皮损的原发性C-ALCL患者的首选;假若皮损已消退或外科切除皮损,无需进一步治疗。目前主张皮损数目多且不消退的病例最好采用放疗或小剂量的氨甲喋呤口服;对于有其他脏器受累的病例,应该采用联合化疗。据研究,多中心皮疹和局部淋巴结受累患者的预后与单一皮疹和无淋巴结受累患者的预后无差别,有细胞间变性的病例与非细胞间变性的病例在临床表现、病程演变及预后方面均无差别[3]。虽然患者自诉20年前即发病后自愈,由于未进行组织病理学检查而无法确诊。本次发病病程约2年,而无自然消退,结合临床表现、组织学特征及免疫表型,诊断为原发性皮肤间变性大细胞淋巴瘤,虽然病程较长,但皮损仍局限于下颌,表明其愈后较好。
[1]李雪,朱文静,金仙花,等.原发性皮肤间变性大细胞淋巴瘤1例[J].中国皮肤性病学杂志,2013,27(2):179-180.
[2]Bolognia JK,Jorizzo JL,Rapini RP.皮肤病学[M].朱学骏,王宝玺,孙建方,等主译.北京:北京大学医学出版社,2014:2285-2308.
[3]黄怀球,张静,钟毅,等.原发性皮肤间变性大细胞淋巴瘤1例并临床分析[J].皮肤性病诊疗学杂志,2012,19(6):365-366.
R733.41
B
1672-0709(2016)06-0379-02
2016-03-02)
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
今日农业(2022年3期)2022-06-05
建材发展导向(2021年14期)2021-08-23
祝您健康·文摘版(2019年4期)2019-06-11
环球时报(2019-04-03)2019-04-03
百科探秘·航空航天(2018年11期)2018-11-29
军事文摘(2017年22期)2017-11-01
饮食科学(2016年3期)2016-07-04
中国医疗美容(2015年2期)2015-07-19
