
一个常染色体显性遗传非综合征型聋家系的听力学及遗传学特征分析△
2016-02-23 08:27牛志杰蒋璐梅凌云冯永陈红胜贺楚峰
听力学及言语疾病杂志 2016年1期
牛志杰 蒋璐# 梅凌云 冯永,3 陈红胜 贺楚峰
刘亚兰1,2 王雪萍1,2 刘畅1,2 熊俊3
一个常染色体显性遗传非综合征型聋家系的听力学及遗传学特征分析△
牛志杰1,2蒋璐1,2#梅凌云1,2冯永1,2,3陈红胜1,2贺楚峰1,2
刘亚兰1,2王雪萍1,2刘畅1,2熊俊3
【摘要】目的分析一个常染色体显性遗传非综合征型聋家系的听力学和遗传学特征。方法对收集到的一个常染色体显性遗传非综合征型聋家系成员进行家系调查、听力学检测和全身体格检查,绘制家系图谱,整理、分析家系成员的听力学和遗传学特征;提取外周血DNA,对已知常见耳聋基因GJB2、GJB3、COCH、EYA4以及线粒体DNA全序列进行筛查。结果该家系由5代53名成员组成,现存4代42人,耳聋患者11人;耳聋表型连续遗传,男女均可患病,符合常染色体显性遗传规律,均表现为对称性语后感音神经性聋(12~36岁之间发病),起初为高频听力下降,随着年龄的增长,逐渐累及中低频听力。已知常见致聋基因全编码序列突变检测分析无阳性发现。结论该常染色体显性遗传非综合征型聋家系中耳聋者表现为对称性、迟发性、进行性、高频下降为主的语后感音神经性聋。
【关键词】常染色体显性遗传;遗传性聋;基因突变;家系
网络出版时间:2015-12-2815:13
网络出版地址:http://www.cnki.net/kcms/detail/42.1391.R.20151228.1513.032.html
Genetic and Audiological Characteristics of An Autosomal Dominant
听力障碍是最常见的感觉缺陷性疾病之一,全世界范围内估计有3.6亿人承受着这种疾病的痛苦(www.who.int),约每500个新生儿中就有1个为耳聋患儿[1]。随着现代医学的进步和发展,环境因素致聋比例逐渐降低,现听力障碍人群中近60%被认为是遗传性因素所导致[2],鉴定所有的致聋基因对人类遗传学来说是一项艰辛而富有挑战性的工作。
遗传性聋中近70%是非综合征型聋(nonsydromic hearing loss, NSHL),其中50%被认为是单基因遗传病,NSHL中约20%为常染色体显性遗传[3]。目前共定位133个非综合征型聋基因座位(http://hereditaryhearingloss.org,截止2015.08.28)。已报道的NSHL致病基因的定位克隆主要是依据家系研究,所以对临床耳聋家系的收集、保存和分析对遗传性聋基因研究很关键。本研究拟通过对收集到的一个常染色体显性遗传性非综合征型聋家系成员的听力表型及遗传特征进行分析,并对其致病基因进行初步筛查,为后续基因定位及克隆研究提供参考。
1资料与方法
1.1家系资料及调查方法家系资料调查由湘雅医院耳鼻咽喉头颈外科家系采集小组完成,调查研究获得湘雅医院伦理委员会认可。该家系5代(53人)居于湖南省某市,现存家系成员42人(22男,20女),其中确诊患者11人(7男,4女),主要调查家族史、耳毒性药物应用史、外伤及手术史、耳聋患者发病史等,家族中共14人(包括11例耳聋者)均接受了全身体格检查、智力评估、耳鼻咽喉科专科检查及听力学评估。根据该家系调查结果,绘制家系遗传图谱之后,依照特异的遗传学规律及临床特点对家系遗传方式进行分析。
1.2听力学检测对所有家系成员均进行常规纯音测听(丹麦Madsen502便携式听力计)、声导抗检测,耳聋患者进行听性脑干反应(ABR)、耳声发射以及40 Hz听觉相关电位检测;先证者耳鸣症状显著,完善耳鸣检测。
根据WHO(1997)的听力损失标准评估听力损失程度,轻度听力损失为26~40 dB HL;中度听力损失为41~60 dB HL;重度听力损失为61~80 dB HL;极重度听力损失为>81 dB HL。
1.3耳聋基因筛查所有家系成员均签署知情同意书,收集耳聋患者血样9份,正常人血样5份(EDTA采血管抽取外周静脉血3~8 ml),用于基因组DNA提取与保存。运用sanger测序技术对先证者进行最常见显性遗传性聋基因GJB2、GJB3、COCH、EYA4以及线粒体DNA[4~6]全序列测序,对于未被报道的新的碱基突变则进一步在部分家系成员中扩大范围筛查与验证,分析是否存在符合相应的基因型和表型共分离现象。
2结果
2.1该家系遗传学特征分析该家系系谱图见图1,可见,家系共5代,各代连续发病,现存家系成员42人,主要为第三至第五代成员确诊的11例耳聋患者,听力损失发病年龄为12~36岁,均以耳部症状为单一症状,无耳毒性相关药物以及噪声接触史,未见明显其他器官、系统异常。耳聋患者中伴耳鸣3例,均无前庭功能障碍。先证者(Ⅳ-17)为27岁女性,因双耳听力下降伴高调耳鸣就诊,纯音测听示双耳对称性、重度感音神经性聋,无眩晕,耳鸣检测示双耳高调性耳鸣,双耳听阈曲线均为高频快速下降型;双耳鼓室导抗图为A型;双耳ABR反应阈为97 dB nHL;DPOAE检查示左耳0.75、1.0、1.5 kHz通过,右耳0.75、1.0 kHz通过,其余频率双耳均未通过,排除了听神经病;颞骨高分辨率CT示:中耳及内耳发育正常,排除中耳乳突病变、内耳畸形和占位性病变等非遗传性因素造成的听力损失。家系中最初的耳聋者为男性(I-1),其后代三个子女中2个为患者,第3代及以后代系患者均系该2名患者的后裔。由此可见,该家系连续四代中均有发病患者,耳聋表型代代相传,呈连续遗传现象;每一代家系成员中男女均可患病,且子代患病个体的父母双方必有一方为耳聋患者; 家系中无论男性或女性患者,均可将耳聋表型遗传给子代,以上特征符合典型的常染色体显性遗传模式。
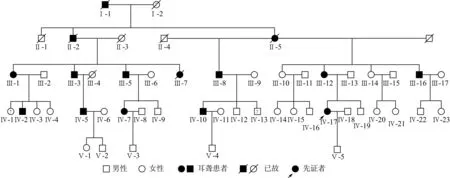
图1 耳聋家系系谱图
2.2听力检测结果该家系11例耳聋患者中年龄最大78岁,最小15岁,其中,第Ⅲ代成员共9人,明确耳聋患者6人,男女之比为4:2,发病年龄最早15岁,最晚32岁,女性患者平均年龄24.5岁,因所有患者平均年龄已达61.2岁,低中频率听力亦受累下降,除了Ⅲ-16患者为重度感音神经性聋,其余均为极重度感音神经性聋,听阈曲线为陡降型和岛状型 (残余听力)[7]。第Ⅳ代成员19人中,耳聋5人,男女之比为3:2,平均年龄41.4岁,最早发病者为12岁,最晚36岁,女性患者平均年龄20.2岁,听阈曲线均以高频听力下降明显,部分患者出现中频听力受累,而低频听力基本均正常。该家系11例耳聋者中10例患者的听力检测及发病年龄、双耳平均听阈、纯音听阈曲线类型、听力损失程度及是否伴耳鸣与否见表1,Ⅳ-10右耳平均听阈虽在正常范围,但高频听力下降明显,且伴耳鸣症状,考虑其发病时间短,表型同先证者相似度高,故初步将其视为患者;Ⅲ-8、Ⅳ-2不愿配合血采样及听力检测,但通过言语交流发现其听力差、言语交流困难,可明确为耳聋患者,第Ⅴ代家系年龄普遍偏小,尚未到发病年龄。
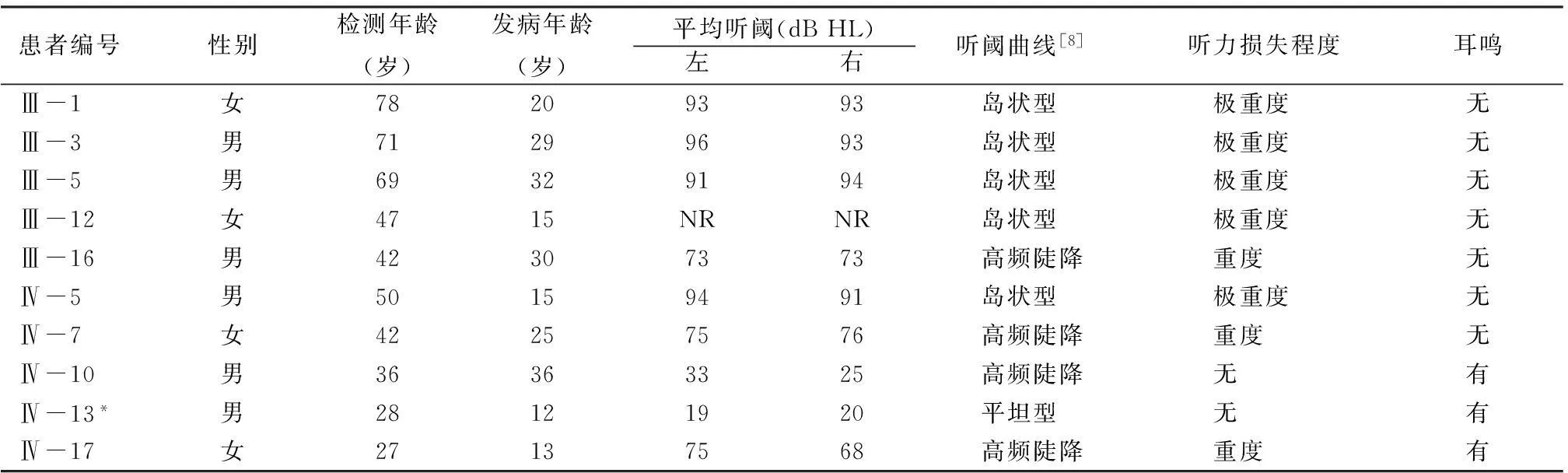
表1 该家系中10例耳聋患者听力检测及发病年龄、纯音听阈测试结果及是否伴耳鸣
注:*尚未到发病年龄或刚到发病年龄表型可疑者; NR为各频率最大输出均无反应
2.3基因突变筛查结果家系中先证者基因直接测序结果同参考序列比对后,发现线粒体基因共32个碱基变异,GJB2基因和GJB3基因各1个碱基变异,EYA4基因共6个碱基变异,COCH基因共3个碱基变异,该4个常见致病基因以及线粒体DNA全编码序列筛查突变位点均为已报道的单核苷酸多态性改变;COCH基因c.629+38Delt未见报道,对家系一例患者(Ⅲ-12)和两例正常者(Ⅲ-13、Ⅲ-14)验证该位点,发现Ⅲ-12、Ⅲ-13都带有该突变,而Ⅲ-14为正常基因型,未发现该碱基改变与临床表型的共分离现象(图2)。
3讨论
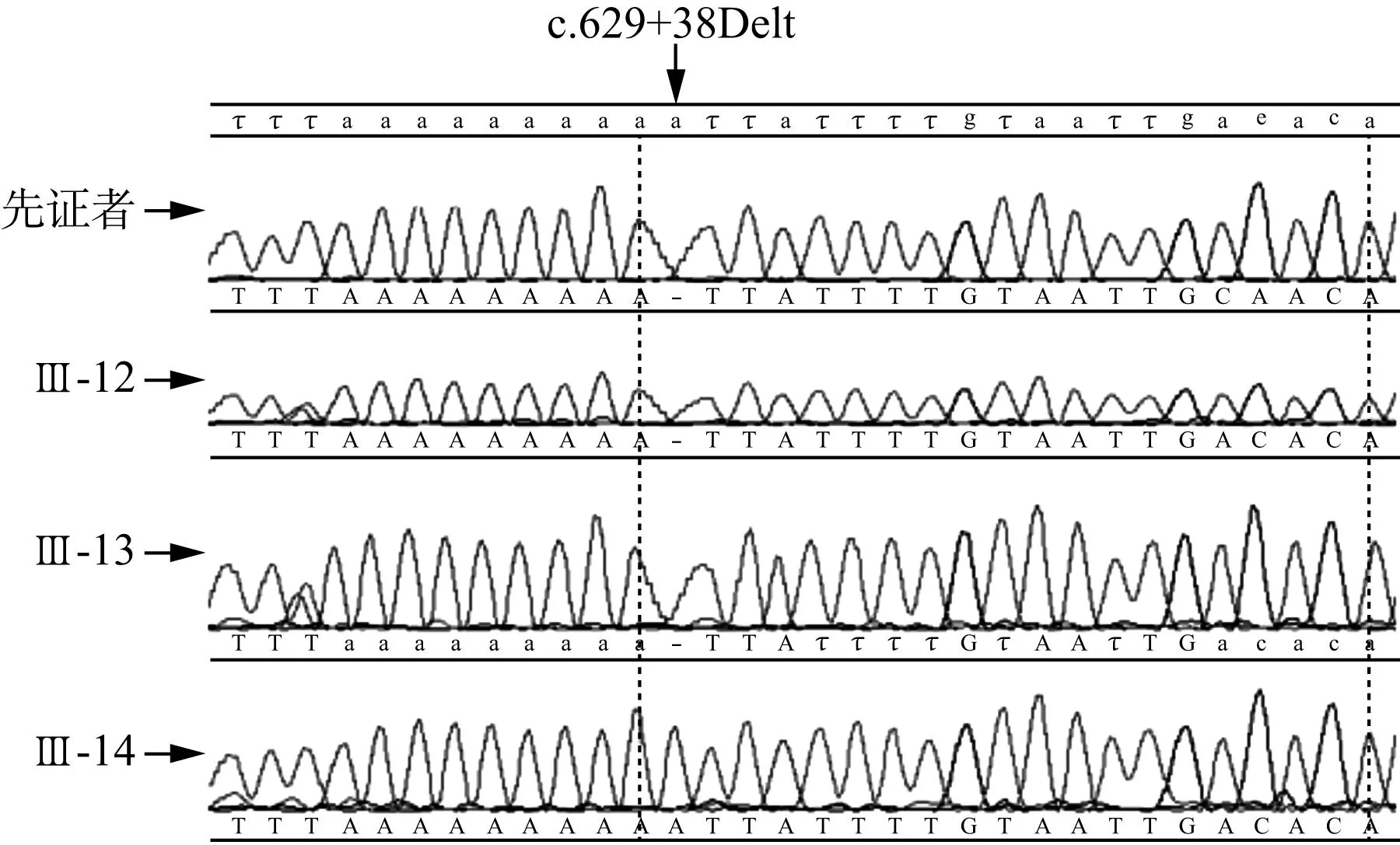
图2 家系成员COCH基因c.629+38Delt位点验证
通过该家系资料调查分析,可见其家系系谱图表现出连续遗传现象, 第三代家系成员中男女患者比例为4:2,但已故的Ⅲ-7为女性,通过家属提供信息判断为耳聋患者,同时家系第四代患病个体男女性别比相近(3:2),由此判断,该家系中男女均可出现耳聋表型,且机率均等,符合典型常染色体显性遗传模式,故可确认该家系为一个常染色体显性遗传性非综合征型聋家系。家系患者均为语后聋,发病年龄12~36岁,起始均为高频听力下降,呈进行性加重,45~50岁以后累及中低频,表现为极重度感音神经性聋,表型特征为语后、迟发型、进行性、高频听力损失为主的感音神经性聋。
常染色体显性遗传性非综合征型聋遗传表型以高频听力损失为主,而低频和中频听力损失表型相对少见,且至今尚未发现某种基因突变频率相对较高的常染色体显性遗传(DFNA),缺乏像常染色体隐性遗传性聋致病基因GJB2一样的“明星基因”,故对高频听力下降为主的DFNA家系致病基因的鉴定工作更为困难。耳聋基因具有极大的异质性,数个基因(GJB2、GJB6、TECTA、MYO7A、MYO6A、COL11A2、TBC1D24以及TMC1等)既可引起显性亦可引起隐性非综合征型聋,还有一些基因(GJB3、GJB2、WFS1、MYO7A、COL11A2以及MYH9等)可引起综合征型和非综合征型聋[9]。遗传性疾病基因定位克隆策略主要有四种方法[10],目前耳聋基因研究多采用的是候选基因克隆和位置候选基因克隆法,该家系样本系2代家系成员,难以提供足够的遗传信息量,因此本研究采用基因直接测序检测突变。
GJB2是最常见的非综合征型聋致病基因,50%的常染色体隐性遗传性聋和20%的遗传性聋与之相关[11],定位于DFNA3A和DFNB1A位点,文献统计GJB2已有超过110种突变方式[12],其中9种可导致DFNA(http://davinci.crg.es/deafness/index.php)。GJB2基因编码的缝隙链接蛋白Cx26通过细胞缝隙链接网络影响内耳钾离子通道的功能[12];GJB2 235delC突变是东南亚人群中发病率最高的突变类型[13];GJB2基因突变表型可表现为轻度到极重度聋[14],患者的运动功能及前庭功能正常[15]。流行病学调查也显示,GJB2基因是我国非综合征型耳聋人群中的热点突变基因,并且该基因仅一个外显子序列,检测方便。
GJB3基因编码缝隙链接蛋白connexin31,被定位和克隆在(DFNA2B)1p33-35,与GJB2(DFNA3&DFNB1)一样被广泛认为可作为遗传性听力损失筛查的良好候选基因[16]。GJB3基因是一种典型的可引起非综合征型聋、综合征型聋及遗传性皮肤病的异质性基因[17,18]。该基因是由中国遗传学重点实验室夏家辉院士等研究人员在两个小的高频听力损失的中国耳聋家系中首先报道[16]。GJB3基因突变引起的耳聋患者,起初多表现为高频听力损失,逐渐累及全频,发病年龄多在10~20岁之间,表型与本研究耳聋家系很相似,并且拥有相同的遗传背景。
COCH基因定位于DFNA9位点[5],DFNA9基因突变所致听力损失患者发病年龄集中于20~30岁,起初表现为高频受累,逐渐累及全频,表现为严重听力缺失,部分患者可伴眩晕等前庭功能障碍[19];COCH基因突变在DFNA家系中检出频率相对较高。EYA4基因突变引起的耳聋主要表现为30岁左右开始出现高频和中频听力损失,逐渐累及全频,听阈曲线为下降型或平坦型[20],本研究收集的耳聋家系患者表型与此类似,故该家系的致病基因可能是EYA基因突变。
线粒体基因突变可以导致非综合征型和综合征型听力损失,其中12SrRNA基因突变还可以引起患者对氨基糖苷类药物的易感性[21]。虽然尚未报道其可引起常染色体显性遗传性聋,但其为热点突变基因,考虑到耳聋基因的异质性,本研究对该家系先证者进行了检测,共发现32个单核苷酸碱多态,初步排除该家系母系遗传的可能性。
根据文中对候选基因筛查的结果,显示COCH基因c.629+38Delt位点经验证未发现基因型和表型的共分离,初步排除该基因为该家系的致病基因,根据序列多物种保守性分析,该位点保守性低,亦可能为新的单核苷酸多态位点,其余GJB2、GJB3、COCH、EYA4以及线粒体DNA共筛查出42个碱基变异,均为已知的单核苷酸多态位点。下一步应该对已克隆的92个NSHL耳聋基因或者33个DFNA基因进行测序筛查(http://hereditaryhearingloss.org,截止2015.08.28),但这是一项非常费时、费力、低效率的工作,且该家系特征限制了全基因组连锁分析方法的应用。
全外显子组测序技术可以进行目标序列的富集,然后对几乎所有的蛋白编码外显子序列进行同步测序,使得全基因序列的获取变得更加便捷,鉴于大多数功能性变异主要位于外显子及其相邻内含子区域,全外显子组测序已成为研究孟德尔遗传性疾病的一种重要技术方法[22,23];近5年应用该技术已经有10余个新的耳聋基因被鉴定。鉴于该新一代测序技术的高效性,今后,拟对该家系成员Ⅲ-14(正常者)、Ⅲ-12、Ⅳ-5进行全外显子组测序,希望能找到该家系的致病基因或新的耳聋基因。
参考文献4
1Morton CC, Nance WE. Newborn hearing screening--a silent revolution[J]. N Engl J Med, 2006, 354:2151.
2Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection[J]. Rev Med Virol, 2007, 17:253.
3Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity[J]. Am J Hum Genet, 1997, 60:758.
4王鸿涵,冯永,王行炜,等. 一个迟发性非综合征型常染色体显性遗传性聋家系表型特征及致病基因初步探讨[J]. 听力学及言语疾病杂志, 2012, 20:309.
5Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics[J]? Mutat Res, 2009, 681:189.
6李丽娜,李庆忠,武文明,等. 散发高频听力下降人群中KCNQ4基因的突变筛查[J]. 听力学及言语疾病杂志, 2007, 15:25.
7顾瑞. 纯音测听[M].见: 姜泗长,闫承先,主编. 现代耳鼻咽喉科学. 天津:天津科学技术出版社, 1994. 11~12.
8王秋菊,译. 关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案[J]. 中华耳科学杂志, 2003, 1:46.
9Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness[J]. Clinical Genetics, 2006, 69:371.
10孙开来,赵彦艳,熊第志,译. 医学遗传学原理[M].北京:科学出版社, 2001.190~197.
11Estivill X, Fortina P, Surrey S, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness[J]. Lancet, 1998, 351:394.
12Angeli S, Lin X, Liu XZ. Genetics of Hearing and Deafness[J]. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology, 2012, 295:1812.
13Yan D, Park HJ, Ouyang XM, et al. Evidence of a founder effect for the 235delC mutation of GJB2 (connexin 26) in east Asians[J]. Hum Genet, 2003, 114:44.
14Liu XZ, Pandya A, Angeli S, et al. Audiological features of GJB2 (connexin 26) deafness[J]. Ear Hear, 2005, 26:361.
15Green GE, Mueller RF, Cohn ES, et al. Audiological manifestations and features of connexin 26 deafness[J]. Audiological Medicine, 2003, 1:5.
16Xia JH, Liu CY, Tang BS, et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment[J]. Nat Genet, 1998, 20:370.
17Lopez-Bigas N, Olive M, Rabionet R, et al. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment[J]. Hum Mol Genet, 2001, 10:947.
18Richard G, Smith LE, Bailey RA, et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis[J]. Nat Genet, 1998, 20:366.
19Robertson NG, Lu L, Heller S, et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction[J]. Nat Genet, 1998, 20:299.
20Wayne S, Robertson NG, DeClau F, et al. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus[J]. Hum Mol Genet, 2001, 10:195.
21Fischel-Ghodsian N, Prezant TR, Chaltraw WE, et al. Mitochondrial gene mutation is a significant predisposing factor in aminoglycoside ototoxicity[J]. Am J Otolaryngol, 1997, 18:173.
22Shearer AE, DeLuca AP, Hildebrand MS, et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing[J]. Proc Natl Acad Sci USA, 2010, 107:21104.
23王洪阳,王秋菊. 目标区域捕获联合新一代测序技术在遗传性聋研究中的应用及发展前景[J]. 听力学及言语疾病杂志, 2014, 22:531.
(2015-01-01收稿)
(本文编辑周涛)
·临床研究·
Nonsyndromic Deafness Kindred
Niu Zhijie*, Jiang Lu, Mei Lingyun, Feng Yong, Cheng Hongsheng, He Chufeng,
Liu Yalan, Wang Xueping, Liu Chang, Xiong Jun
(*Department of Otolaryngology-Head and Neck Surgery, Xiangya Hospital,
Central South University, Changsha, 410008,China)
【Abstract】ObjectiveTo investigate the audiological and genetical features of a large Chinese pedigree with autosomal dominant nonsydromic hearing loss.MethodsWe collected the detailed medical history information of all the family members. Otoscopies, physical examinations, and pure-tone audiometry were performed. Blood samples were collected and well preserved as consented by all the participants. Genetic characteristics of the family were evaluated by audiology materials and pedigree; genomic DNA was isolated from the peripheral leukocytes of the subjects. The common deafness genes (GJB2, GJB3, COCH, EYA4,MTDNA) were screened to exclude known pathogenic mutations in the candidate genes.ResultsWe collected data from a five-generation family from Hunan Province of China with autosomal dominant sensorineural hearing loss. The family had 53 members, of which 42 members were alive and 14 members' blood samples were collected, and 11 patients were found to be hearing-impaired. Audiograms showed bilateral symmetric, progressive and sensorineural hearing loss occurred in the first to third
△国家重大科学研究计划项目(Grant No.2014CB943003)、国家自然科学基金(Grant No.81300833)及湖南省自然科学基金青年基金(Grant No.13JJ4023)资助资助
1中南大学湘雅医院耳鼻咽喉头颈外科(长沙410008);2耳鼻咽喉重大疾病研究湖南省重点实验室;3中南大学医学遗传学国家重点实验室
#为并列第一作者
decade ago, progressing first at high-frequencies, and ultimately expending to mid and low frequencies, leading to severe-profound hearing loss. The mode of inheritance appeared to be autosomal dominant based upon the pedigree. By screening the common deafness genes, we didn't find any causative mutations but some single nucleotide polymorphisms(SNPs).ConclusionThe pedigree analysis indicated an autosomal dominant inheritance pattern in the large family, in which affected subjects showed post-lingual, symmetry, progressive hearing impairment. The negative screening results of the common deafness genes suggest that we should perform other strategies to explore the pathogenic genes, such as applying facilitate whole-exome sequencing or whole genome sequencing on this family.
【Key words】Autosomal dominant hereditary;Hereditary deafness;Gene mutation;Pedigree
通讯作者:梅凌云(Email:entmly@163.com);冯永(Email:fengyonghn@hotmail.com)
作者简介:牛志杰,男,湖南人,在读博士研究生,主要研究方向为遗传性聋为主的耳科学基础与临床研究。
【中图分类号】R764.44
【文献标识码】A
【文章编号】1006-7299(2016)01-0005-05
DOI:10.3969/j.issn.1006-7299.2016.01.002
猜你喜欢
广西林业科学(2022年6期)2023-01-16
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国生殖健康(2020年2期)2021-01-18
中南林业科技大学学报(2019年4期)2019-04-08
小学生导刊(2018年13期)2018-06-29
森林工程(2018年1期)2018-05-14
中国生殖健康(2018年2期)2018-01-12
中国现代医学杂志(2015年26期)2015-12-23
郑州大学学报(医学版)(2015年2期)2015-02-27
