基于DNA弯曲度的H2A.Z核小体定位与修饰研究
2016-01-15 01:37:46单增辉,丰继华,陈攀峰等
生物信息学 2015年1期
基于DNA弯曲度的H2A.Z核小体定位与修饰研究
单增辉,丰继华*,陈攀峰,魏恨恨,胡焕
(云南民族大学电气信息工程学院,昆明 650500)
摘要:在真核生物染色质中,H2A.Z是高度保守的组蛋白变异体, 与转录调控、基因组的稳定性密切相关。为了探讨组蛋白修饰、DNA弯曲度与H2A.Z核小体定位三者之间的关联,在得到实验所测的相关数据后,利用MINE算法并结合皮尔逊相关系数在酵母全基因组的转录起始位点周围探讨了三者间的线性与非线性关系。其中MIC算法可以定量的得出数据之间关联度大小的值,用于衡量数据之间是否存在着关联,而皮尔逊相关系数则用于检查是否为线性关联。结果除了发现大部分组蛋白修饰种类和核小体定位之间存在着线性关联外,还探测到有两种组蛋白修饰数据(H4ac修饰与GCN4修饰)和核小体定位数据之间存在着以往未发现的非线性关系(大致呈正余弦函数),并从数据的生物背景(组蛋白修饰与核小体位置)上探讨了出现非线性现象的原因。
关键词:转录起始位点(TSS); 组蛋白变异体; H2A.Z; 修饰; 核小体
中图分类号:Q-3文献标志码:A
收稿日期:2015-01-28;修回日期:2015-03-01
基金项目:科技部技术开发研究专项(2013EG111232);农业部甜菜产业技术体系项目(CARS-21010202)。
作者简介:龚攀,男,硕士研究生,研究方向:植物生物技术; E-mail:gp68x@126.com.
doi:10.3969/j.issn.1672-5565.2015.01.04
Based on the DNA bending H2A. Z nucleosome positioning and modification research
SHAN Zenghui,FENG Jihua*, CHEN Panfeng, WEI Henhen, HU Huan
(SchoolofElectricalandInformationTechnology,YunnanUniversityofNationalities,Kunming650500,China)
Abstract:In eukaryotes chromatin, H2A.Z is highly conservative histone variants and closely associated with the transcriptional regulation and the stability of the genome and of high importance. In order to explore the links among the histone modification, DNA bending and H2A.Z nucleosome positioning. After getting the relevant data sets, we discussed the linear and nonlinear relationships between those datas around transcription start site in the yeast genome-wide by MIC algorithm and Pearson correlation coefficient. The MIC algorithm got a correlation value to quantificationally measure whether there is an association between datas, while pearson correlation coefficient is used to check whether the correlation is linear. The results showed most of the types of histone modification were linear correlation between the nucleosome positioning. In addition, two kinds of histone modification datas (H4ac modification with GCN4 modification) were found between nucleosome positioning(roughly is positively cosine function) and discussed the reasons of nonlinear phenomena from the biological background(histone modification and nucleosome position) of datas.
Keywords:TSS; Histone variants; H2A.Z; Modify; Nucleosome
真核生物中, DNA和组蛋白结合在一起形成染色体, 核小体是染色体组成的基本结构单位,它含有一个核心组蛋白八聚体结构,该结构由4种组蛋白H2A、H2B、H3和H4组成, 每一种组蛋白各由两个分子形成, 约200 bp的DNA分子缠绕在核心组蛋白八聚体外面形成一个核小体单位[1-3]。核小体还可以形成更高级的染色体结构,因此其位置对基因物质的形成与维护有着重要影响[4]。
组蛋白修饰是在相关修饰酶的作用下发生在核小体组蛋白N末端的共价修饰,这些共价修饰包括甲基化、乙酰化和磷酸化等,不同的组蛋白修饰在基因的表达中起着不同的作用。核小体在基因组 DNA 分子上的精确位置称为核小体定位,核小体定位已被证实在诸如转录调控、DNA 复制和修复等多种细胞过程中起着重要作用。而基因组上核小体位置的确定涉及 DNA、转录因子、组蛋白修饰酶和染色质重塑复合体之间的相互作用[5-6]。DNA物理特性是指DNA链的弯曲度、内在曲率、柔韧性、相邻碱基对的倾斜度等等。研究表明DNA链的物理特征与其他调控因素、化学修饰一起共同调节了真核生物的转录过程[7-10]。
据此,在实验获得的酵母组蛋白变异体H2A.Z核小体定位数据、组蛋白修饰数据、DNA弯曲度数据基础上,分别研究组蛋白修饰数据与核小体定位数据之间的关联,弯曲度数据与核小体定位数据的关联。与以往的研究不同,本文除了研究数据集间的线性关联之外,还借助MIC算法对非线性关联也进行了探讨。
1数据与方法
1.1数据来源
本文研究的数据主要来源于以下几个方面:一是Julia Zeitlinger等人测定的酵母中核小体组蛋白甲基化及乙酰化修饰的数据[4]。二是Luc Gaudreau等人所测的关于组蛋白变异体H2A.Z的核小体定位数据[17]。三是通过查阅数据库所得的DNA弯曲度数据。由于实验测得的原始数据格式和精度不统一,所以对原始数据进行了必要的预处理。
1.2数据预处理
1.2.1数据插值
为了将各种数据统一为1 bp精度,本文首先对各组数据进行插值,在综合比较几种常见的插值方法后,我们在对插值后形成的图谱(对齐后)与文献[11]的研究结果进行对比过程中,发现使用高斯插值方法效果较好。
1.2.2数据对齐
根据基因的位置数据,在每个基因的TSS周围,选取上、下游各 1 200 bp的长度范围(经过反复尝试、对比发现该长度研究效果最好),分别对核小体定位数据,组蛋白修饰数据,DNA弯曲度数据进行数据截取与对齐处理(其中C型基因对应的数据做了反转处理),再将以上数据叠加平均并做了归一化处理,由此得到全基因组在TSS附近归一化后的核小体定位图谱、组蛋白修饰图谱以及弯曲度图谱。
通过把插值对齐后的图谱(见图1)和Yuan GC等人实验测得的数据相比较发现[1],数据分布及走势是一致的,因此可以看出用高斯插值所得到的全基因组数据是正确的。并且从以上图谱可以看出各组数据在TSS附近的分布呈现出一定规律,如H3.H2O2组蛋白修饰在TSS处于低谷。其中组蛋白修饰数据(甲基化与乙酰化)有28个图谱,因篇幅所限,本文仅列举了H3.H2O2的修饰图谱。
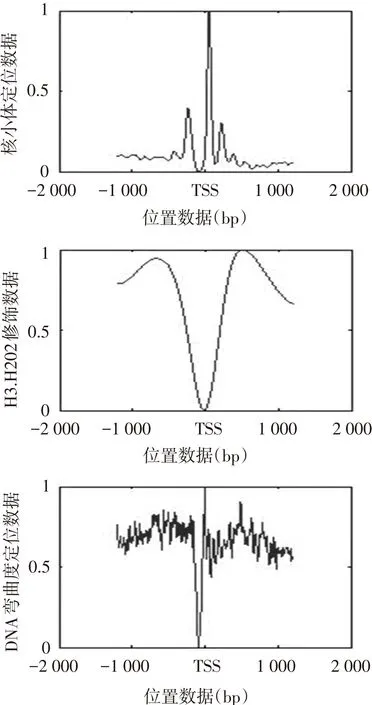
图1 在TSS附近对齐的核小体定位、
1.3MIC算法
MIC算法是一个研究数据之间关联度的新算法,在这里就其主要原理进行介绍。MIC是用来测量两变量依赖关系的算法,它能够捕捉到两变量之间广泛的关联,包括函数与非函数关系(包括变量间原线性与非线性关系)。并且对于函数关系,可以得到一个大致等于样本判定系数的值,它属于基于非参数检测统计量最大化信息方法中的一大类。其大致思想是:如果两个变量之间存在关系,那么首先在这两个变量的散点图上绘制出一个网格,对数据进行分区以封装其关联。然后计算两组数据之间的MIC值,通过探索所有的网格至其最大的网格分辨率,然后再根据样本大小计算每一个整数对(x,y)的最大交互信息,之后把这些交互信息值归一化,最后将每组归一化后的最大交互信息值组成一个矩阵——特征矩阵M。而MIC的值就是特征矩阵M的最大值。
其主要公式如下:
对于一有限定义集D⊂R2与整数x,y定义:
(1)
其中:x代表列,y代表行,I(D|G)代表(D|G)的交互信息
(2)
特征矩阵M(D)为:
(3)
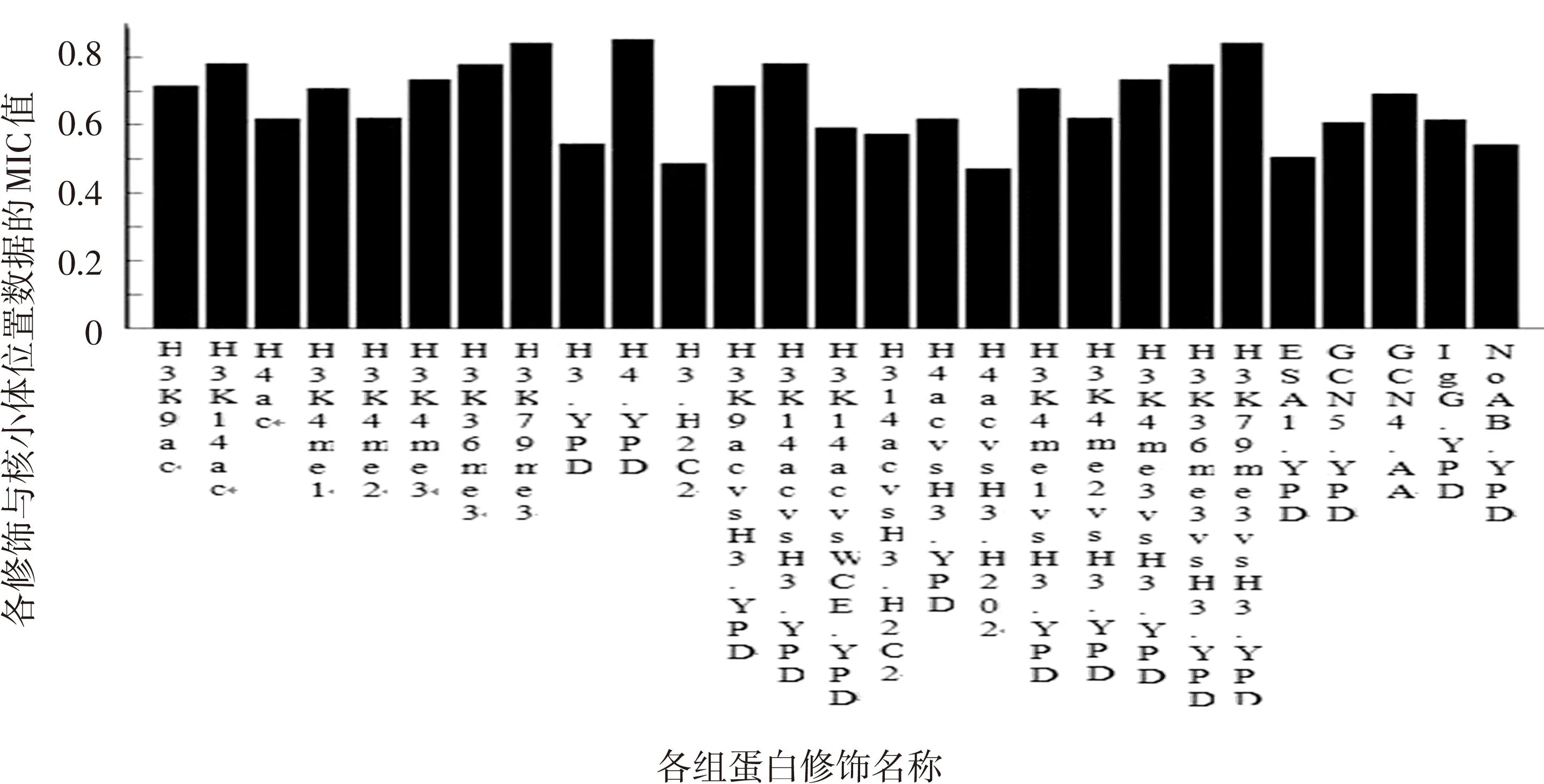
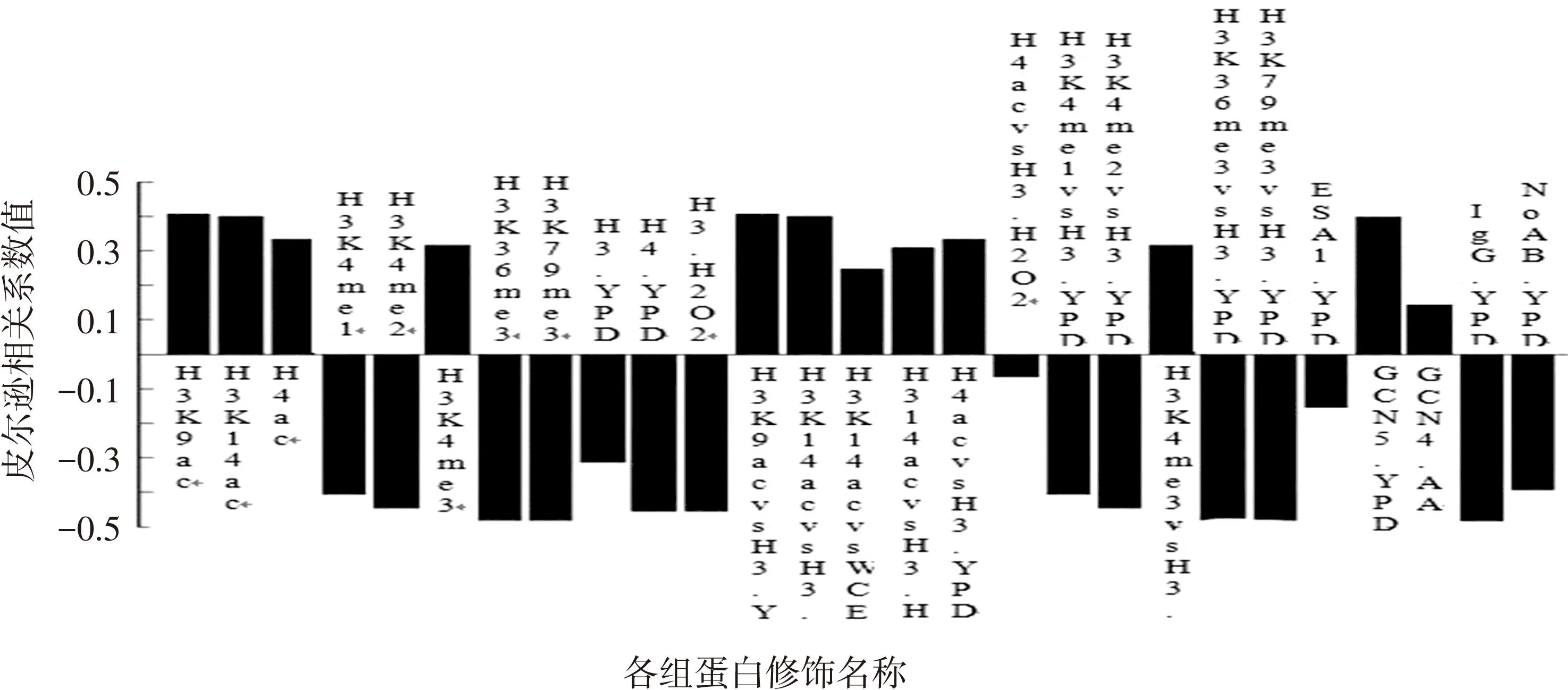
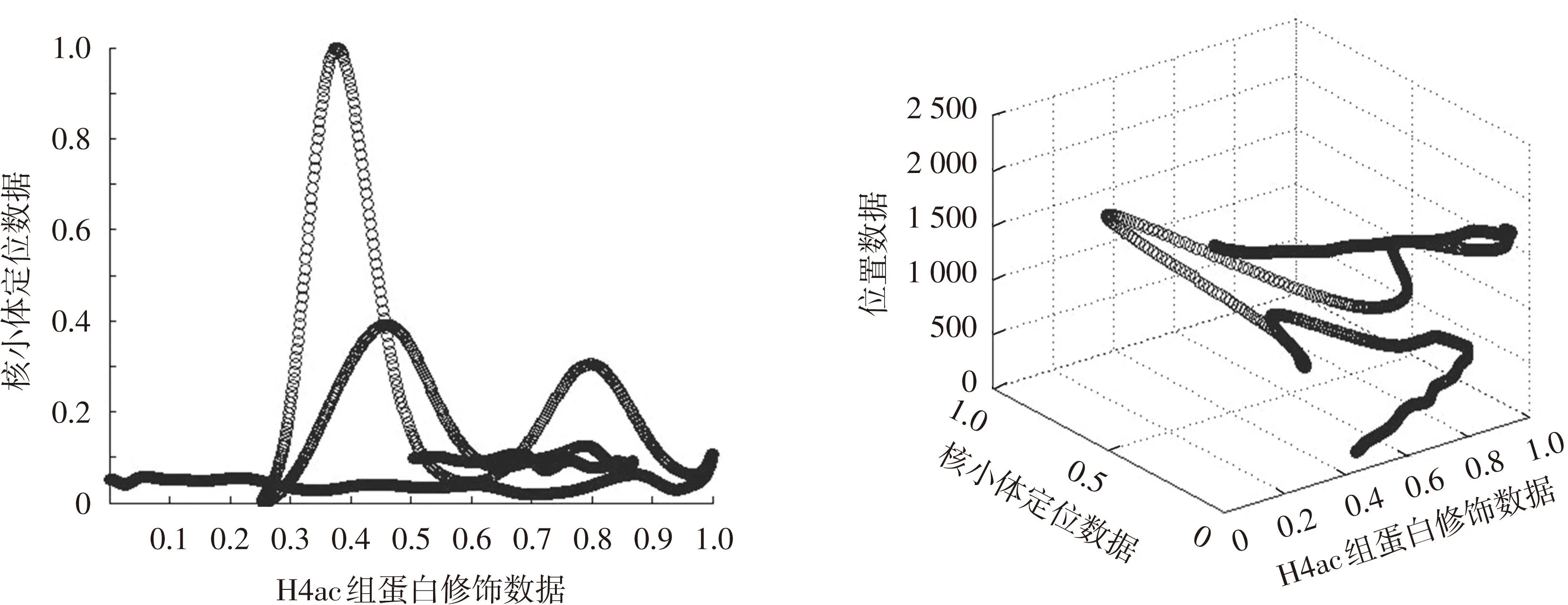
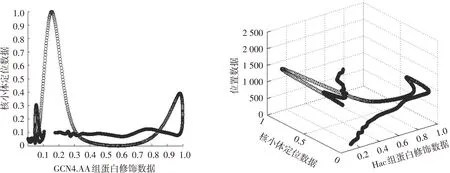
而在xy (4) 其中n代表样本大小,B(n)代表网络大小,通常B=n0.6,MIC(D)表示最大信息系数。 2结果 2.1MIC值的定量关联性探究 基于以上的综合介绍(数据的预处理与算法),为研究组蛋白修饰与核小体定位的关系,本文首先以MIC算法为基础,计算出两者数据之间关联度,并得到以下条形图(见图2)。 图2 各修饰数据与核小体定位数据MIC值的条形图 2.2皮尔逊相关系数的线性探究 由上图表MIC值可以说明数据之间有着较强的关联性。但这种关联究竟是线性关联还是非线性呢?为此,又计算了数据间的皮尔逊相关系数,得到核小体占位数据分别与27种修饰数据的皮尔逊相关系数图表(见图3)。 图3 各修饰数据与核小体定位数据的皮尔逊相关系数条形图 通过仔细的对比了两种不同方法下的数据,发现大部分数据符合实际。如MIC 值高其皮尔逊相关系数也比较高(如第一种修饰H3K9ac),当两个MIC值想接近时,其对应的皮尔逊相关系数也接近(如第一种修饰H3K9ac和第四种修饰H3K4me1),MIC值比价小时皮尔逊相关系数比较小(第23种修饰ESA1.YPD)。但仔细对照可以发现其中有两组数据差异很明显,第17种修饰H4ac与第25种修饰GCN4.AA,它们的MIC值较高而皮尔逊相关系数确很低,所以在此猜想这两种修饰之间可能存在着未知的非线性关系。 2.3数据走势图的非线性探究 基于此,本文在全基因组下对TSS附近的归一化的核小体定位数据与归一化的该两种修饰数据分别作二维与三维图(把位置信息添加进去),结果如图4、图5所示。 图4 H4ac修饰与核小体定位数据的二维与三维图 图5 GCN4.AA修饰与核小体定位数据的二维与三维图 从图4和图5中可以发现图形的二维投影走势呈正余弦函数(局部更明显),并且其极值均处于TSS(1 201点处)位置左右。通过查阅资料发现在酵母生物体中组蛋白H4的乙酰化发生在组蛋白尾部几个不同的赖氨酸位置上[13-15],其乙酰化高峰期超过了活跃基因的开始位置并且与转录速率、转录积极性有关,而且不能排除H4的N尾端个别赖氨酸残留物的乙酰化对转录活动也许有着不同的关联,所以猜想是因此造成了如图所示的非线性关系。而对于GCN4本文依据推测上图是由于GCN4基因在氨基酸控制脱抑制反应中所需求[16],GCN4蛋白在基因5端未翻译的区域中保护着重复的区域,而在GCN4区域某些位置处选择性对启动子的约束是和这些点与GCN4的相对亲和力有关而造成的。 而后用同样的方法对核小体定位数据与DNA弯曲度数据进行了探讨,发现两者之间的MIC值和皮尔逊相关系数均比较小,其中MIC值为0.325 44,皮尔逊相关系数为0.039。通过查阅文献发现[8],尽管核小体的定位与DNA物理特性又有着很大的关联,但在体内,决定核小体位置的因素很多,加上数据的误差等,导致了两者间关联性并不大。 4结语 本文在得到核小体定位数据、组蛋白修饰数据和弯曲度数据的基础上,综合比较了几种常见的插值方法,并把插值后的图谱与前人的研究进行对比,最后确定了利用高斯方法进行插值,然后对数据进行对齐和归一化处理,最终得到了全基因组下TSS附近的各类数据与图谱。然后利用MINE算法计算了数据之间的关联度,结果发现MIC值均比较大,这说明数据之间有着很强的关联,为了明确这种关联之间是否存在非线性,我们又计算了数据间的皮尔逊相关系数,结果发现大部分组蛋白修饰与核小体定位数据之间的皮尔逊相关系数值都很高,存在着很强的线性关联。但还发现出有两种修饰H4ac与GCN4和核小体定位数据之间的MIC值很高但皮尔逊相关系数值确很小,为了探测两者间是否为非线性关联,本文又结合了两个修饰种类与核小体数据间的二维及三维走势图,最终发现了两者之间存在的非线性关系。 参考文献(References) [1]KOUZARIDES T. Chromatin modifications and their function[J].Cell, 2007, 128(4): 693-705. [2]ZHANG Z, PUGH B F. High-resolution genome-wide mapping of the primary structure of chromatin[J].Cell , 2011, 144(2):175-186. [3]XING Yongqiang, LIU Guoqing, ZHAO Xiujuan, et al. An analysis and prediction of nucleosome positioning based on information content [J]. Chromosome Research, 2013,21 (1):63-74. [4]JULIA Z ,FRAN L, RICHARD A. Genome-wide map of nucleosome acetylation and methylation in yeast[J]. Cell, 2005, 122: 517-527. [5]CUI Xiangjun, LI Hong. Advances on the combinatorial patterns of histone modifications[J]. Journal of Inner Mongolia University (Natural Science Edition), 2012, 43: 101-111. [6]WANG Jianying, WANG Jingyan, LIU Guoqing. Calculation of nucleosomal DNA deformation energy: its implication for nucleosome positioning[J]. Chromosome Research, 2012, 20 (7): 889-902. [7]LIU Hui, ZHUANG Ziheng, GUAN Jihong, et al. Transcriptional regulation functions of nucleosome positioning: a survey[J]. Progress in Biochemistry and Biophysics,2012, 39: 843-852. [8]VINCENT M, CEDRIC V. DNA physical properties determine nucleosome occupancy from yeast to fly[J]. Nucleic Acids Research, 2008,36(11):3746-3756. [9]ITAY T,JUDITH B,NAAMA B. The pattern and evolution of yeast promoter bendability[J]. Trends in Genetics, 2007,23(7):318-321. [10]CAI Lu, LUO Liaofu. The DNA of the bending and its topology [J]. Journal of Baotou Iron and Steel Institute,1998,17(2):161-167. [11]YUAN G C, LIU YJ, DION M F,et al. Genome-scale identification of nucleosome positions in S.cerevisiae[J]. Science , 2005, 309 (5734): 626-630. [12]TERRY S. A correlation for the 21stcentury[J].Science,2011,334:1502-1503. [13]XING Yongqiang, LIU Guoqing, ZHAO Xiujuan, et al. An analysis and prediction of nucleosome positioning based on information content[J]. Chromosome Research,2013, 21:63-74. [14]DAVID N, YAKIR A R, HILARY K F et al. Detecting novel associations in large data sets[J]. Science, 2011,334: 1518-1524. [15]PHAM T, TRAN D. Qualitatively predicting acetylation and methylation areas in DNA sequences[J]. Genome Informatics, 2005, 16(2): 3-11. [18]GERALD R. GCN4 protein, a positive transcription factor in yeast, binds general control promoters at all 5′TGACTC 3′sequences[J]. Natural,1986, 83: 8516-8520. [17]PECKHAM H E, THURMAN R E, FU Y,et al. Nucleosome positioning signals in genomic DNA[J]. Genome Res, 2007, 17(8): 1170-1177. *通信作者:崔杰,博士,副教授,研究方向:植物抗逆分子机制;E-mail:cuijie2006@163.com.