
影响伊立替康疗效和毒性的遗传因素
2016-01-13 03:04赵仁永,张蕊,郭瑞臣
药学研究 2015年1期
关键词:多态性
影响伊立替康疗效和毒性的遗传因素
赵仁永1,2,张蕊3,郭瑞臣3
(1.山东大学药学院,山东 济南 250012;2.山东省医药工业研究所,山东 济南 250101;
3.山东大学齐鲁医院,山东 济南 250012)
摘要:伊立替康是一种半合成的喜树碱衍生物,广泛用于多种肿瘤的化疗。伊立替康体内处置过程复杂,药动学和药效学个体差异大,涉及众多多态性药物代谢酶和药物转运蛋白,如羧酸酯酶、尿苷二磷酸葡萄糖醛酸基转移酶、细胞色素P450 3A酶、β-葡萄糖醛酸酶、孕烷X受体、ATP-依赖性转运蛋白等。本文对这些影响因素及其基因多态性的研究进展进行了综述。
关键词:伊立替康;多态性;药物代谢酶;药物转运蛋白
作者简介:赵仁永,男,主管药师,研究方向:新药研发方面的信息情报,E-mail:xiaoyaolaozhao@163.com
中图分类号:R979.1文献标识码:A
The genetic factors of impacting irinotecan efficacy and adverse reactions
ZHAORen-yong1,2,ZHANGRui3,GUORui-chen3
(1.SchoolofPharmaceuticalSciences,ShandongUniversity,Jinan250012,China;2.Shandong
InstituteofPharmaceuticalIndustry,Jinan250101,China;3.QiluHospitalof
ShandongUniversity,Jinan250012,China)
Abstract:Irinotecan is a semisynthetic derivant of camptothecin,is widely used for chemotherapy of a variety of tumors.The pharmacokinetics of irinotecan is extremely complex and the individual variation is significant.Irinotecan′s metabolism is affected by various polymorphic enzymes and drug transporters,including carboxylesterases,uridine diphosphate glucuronosyltransferase,cytochrome P450 3A,β-glucuronidase,pregnane X receptor and the adenosine-triphosphate dependencing transporters.In this article,these factors and the gene polymorphism were reviewed.
Key words:Irinotecan;Polymorphism;Enzymes;Transporter
伊立替康(irinotecan,CPT-11)是半合成喜树碱衍生物,为选择性拓扑异构酶Ⅰ(Topo Ⅰ)抑制剂。化学名(S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14- dioxo1H-pyrano[3′,4′:6,7]-indolizino[1,2-b]quinolin-9-yl-[1,4′bipiperidine]-1′-carboxylate,分子式:C33H38N4O6。
CPT-11由养乐多本社、第一三共、罗纳普朗克罗尔(后更名为安万特,即赛诺菲·安万特)和法玛西亚(辉瑞公司)共同研发,1994年于日本上市,作为二线药物用于5-氟尿嘧啶(5-fluorouracil,5-FU)治疗后复发的结直肠癌患者。美国FDA 1996年批准上市,申报企业为法玛西亚普强。目前为止,其批准适应证包括非小细胞肺癌、胃肿瘤、胰腺肿瘤、非霍奇金淋巴瘤、转移性结直肠癌、宫颈肿瘤、头颈部肿瘤、子宫肿瘤、肺肿瘤、脑肿瘤、结肠肿瘤、卵巢肿瘤、结直肠肿瘤等。
1伊立替康药理作用与体内处置过程
1.1伊立替康药理作用CPT-11在体内经羧酸酯酶(Carboxylesterases,CES)催化,脱去C10位基团,代谢成7-乙基-10-羟喜树碱(SN-38),其抗癌活性为CPT-11的100~1 000倍[1]。
CPT-11(或SN-38)通过抑制Topo Ⅰ发挥其抗肿瘤作用。拓扑异构酶广泛存在于生物体内,参与DNA复制、转录、重组、修复等关键的核内过程。Topo Ⅰ抑制剂并不直接抑制Topo Ⅰ的催化活性,而是通过阻断与DNA反应的最后一步,即DNA在切口部位的重新结合而发挥疗效。Topo Ⅰ抑制剂进入细胞后与拓扑异构酶-DNA复合物共价结合,形成拓扑异构酶-抑制剂-DNA复合物,并稳定这一复合体,导致DNA重连步骤被抑制, DNA链断裂, DNA复制和RNA合成受阻,进而出现细胞凋亡,抑制细胞分裂。
1.2伊立替康体内处置过程CPT-11的体内处置过程表现为二室或三室模型,与剂量和给药方案无关,也不因合并用药而发生改变。CPT-11达峰浓度(Cmax)和药时曲线下面积(AUC)随剂量增加按比例线性增加,表现为线性药代动力学特征[2]。CPT-11输注结束即可达其血浓度峰值,活性代谢产物SN-38达峰浓度时间为0.5~3.75 h。输注结束0.5~1.0 h,血中可测到CPT-11及SN-38反弹浓度,提示经胆汁排泄进入肠道后可被重新吸收进入血液,存在肠肝循环[3]。CPT-11终末相半衰期为14.2 h,血浆清除率为15 L·h-1·m-2,稳态分布容积为157 L·m-2,表明体内广泛分布。此外,胸膜腔液、胆汁、汗液和唾液也可测到高浓度CPT-11[4]。SN-38有与CPT-11平行的血浆分布,半衰期为13.8 h。
动物和人体CPT-11的消除方式主要为肝脏代谢和胆汁分泌。其代谢酶包括CES、尿苷二磷酸葡萄糖醛酸基转移酶(UDP-glucuronyl transferase,UGT)、细胞色素P450 3A(CYP3A)和β-葡萄糖醛酸酶(β-glucuronidase,β-G)。经CES代谢转化为活性代谢产物SN-38,或经CYP3A代谢为无活性的氧化产物7-乙基-10-[4-N-(5-氨基戊酸)-1-哌啶基]-羰基喜树碱(APC)和7-乙基-10-(4-氨基-1-哌啶基)-羰基喜树碱(NPC)等,而NPC又几乎可完全经CES水解为SN-38。SN-38经UGT灭活,转化为二级代谢产物SN-38葡萄糖醛酸苷(SN-38G),SN-38G在肠道细菌产生的β-G作用下,又水解为活性产物SN-38。CPT-11的体内代谢过程见图1。
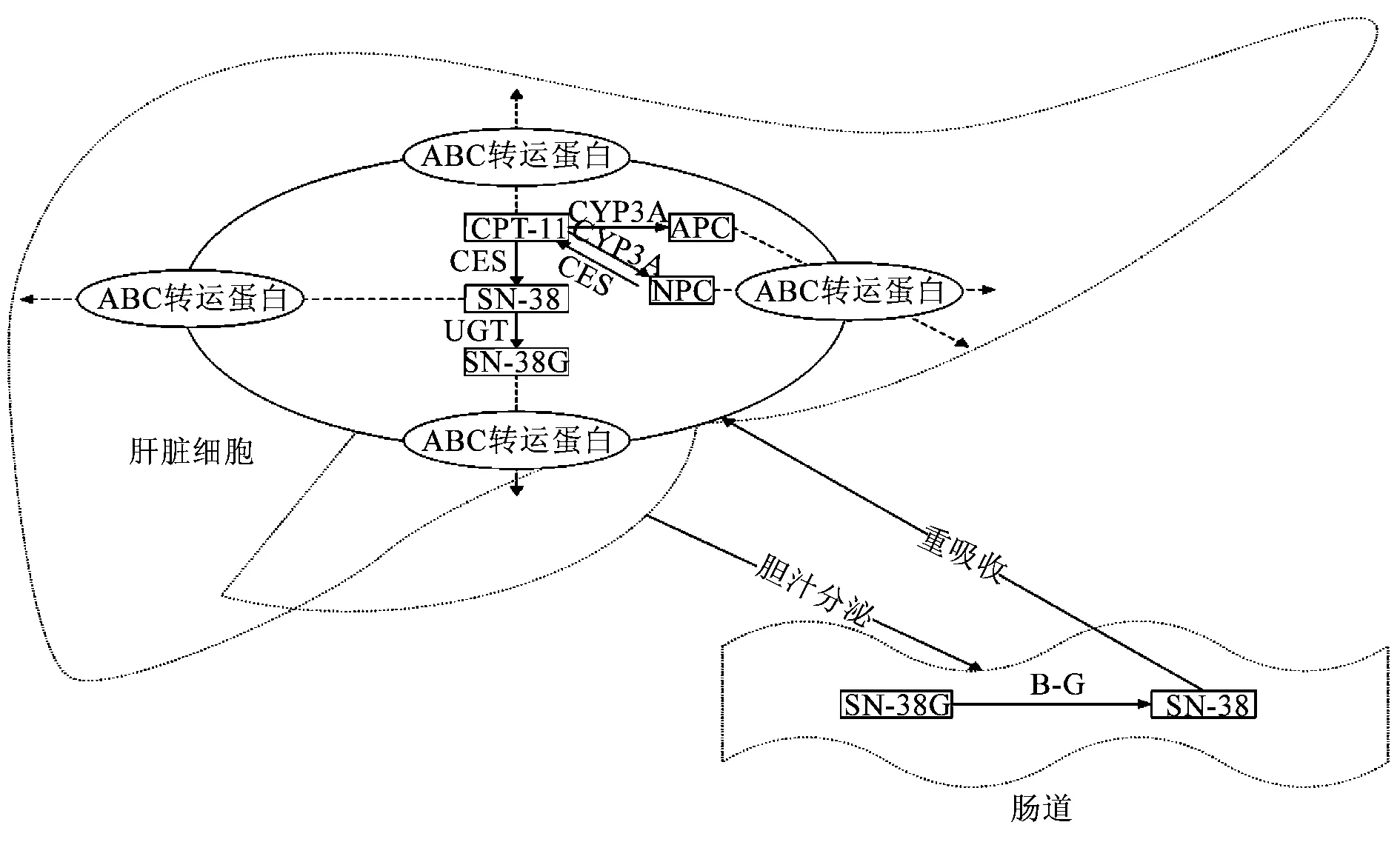
图1 CPT-11的体内代谢过程
细胞内CPT-11、SN-38及其他代谢物由ATP-结合盒(ATP-binding cassette,ABC)转运蛋白转运至细胞外,经肝胆通路从粪便排泄,少部分由尿液排出体外。
2影响伊立替康效应的代谢酶和转运蛋白
已知与伊立替康及其活性代谢物SN-38吸收、代谢、分布、消除密切相关的CES、UGT、CYP3A、β-G、孕烷X受体(PXR)酶及ABC转运蛋白遗传多态性影响伊立替康及其活性代谢物SN-38的体内处置过程,导致效应改变,产生致命性毒性和不良反应,影响伊立替康治疗。
2.1羧酸酯酶(CES)人体存在2种羧酸酯酶,即CES1和CES2,均可使CPT-11代谢为SN-38,但活性不同。Humerickhouse等[5]选择人喉癌上皮细胞,将CES1和CES2分别与CPT-11同时孵育,CES1抑瘤率显著高于CES2,从细胞学角度提示CES1可能主要参与人体CPT-11的转化。Kim SR等[6]对日本人的研究,也未发现CES2对CPT-11体内处置过程有显著影响。CES1于肝脏有较高表达,为CPT-11代谢的主要器官。因此,肝脏CES1表达或活性的改变,如CES1发生突变,存在基因多态性,则可影响血浆中CPT-11代谢物SN-38的浓度,从而影响CPT-11的治疗。此外,Tanimoto等[7]研究表明,CES1基因的mRNA表达与CPT-11对肿瘤细胞的敏感性相关,CES1过度表达可促进CPT-11到SN-38的转化。因此,有人认为CES1基因是预测CPT-11化疗敏感性的因子。
CES1定位于人类17号染色体,存在20多种突变。常见CES1-75G>T突变,分为G/G野生型、G/T杂合子和T/T纯和子突变型。Sai K等[8]对日本人群CES1基因突变概率较高的-75G>T、-30G>A、-186A>C的研究发现,CES1-75G>T突变与CPT-11的生物利用度相关(P<0.05),与(SN-38+SN-38G)/CPT-11]相关(P=0.027)。周喜等[9]对2010年1月~2011年11月接受国产CPT-11联合氟尿嘧啶和四氢叶酸钙(FOLFIRI方案)化疗的晚期结直肠癌患者的研究发现,98例癌患者84例存在基因突变,突变概率为85.7%,其中杂合突变53例,纯和突变31例。CES1-75G/G野生型患者疾病控制率及无进展生存时间均显著高于G/T+T/T突变患者(P<0.05),化疗后KPS评分稳定+好转的比例明显高于G/T+T/T突变患者(P<0.05),但两组患者临床有效率无明显差异(P>0.05)。CES1-75G>T突变与CPT-11毒副反应的发生率无显著相关性。因此,进行CES1基因检测,G/G基因型患者,建议使用含CPT-11的化疗方案;G/T或T/T突变基因型患者,建议改换其他方案取代CPT-11,有助于提高疗效,避免医疗资源浪费。
2.2尿苷二磷酸葡萄糖醛酸基转移酶(UGT)UGT属Ⅱ相代谢酶。人类UGT家族可分为参与酚和胆红素代谢的UGT1家族和参与类固醇代谢的UGT2家族,UGT1家族进一步分为UGT1A、UGT2A和UGT2B三个亚型。UGT1A1、UGT1A7和UGT1A9参与伊立替康活性产物SN-38的葡萄糖醛酸化过程。
UGT1A1主要分布于肝脏,以尿苷二磷酸葡萄糖醛酸(UGA)为糖基供体,与底物发生结合反应,增加底物的水溶性,从而使底物易于溶解,随胆汁或尿液排出体外。UGT1A1可修饰、改变代谢物活性,使代谢物失活,是机体重要的解毒过程。UGT1A1基因多态性主要来自启动子区TATA盒的变异,变异范围包括5~8个TA重复片断。白种人发现4种等位基因(TA)5、(TA)6、(TA)7、(TA)8,而亚洲人只发现(TA)6和(TA)7,组合成TA6/6野生型(UGT1A1*1/*1)、TA6/7杂合子(UGT1A1*1/*28)和TA7/7纯合子(UGT1A1*28/*28)。其中突变型(TA6/7和TA7/7)SN-38葡萄糖醛酸化活性比野生型(TA6/6)低,有更多SN-38蓄积,从而导致急性腹泻(用药24 h以内)和迟发性腹泻(用药24 h以后)、胆碱能综合征和血液学(中性粒细胞减少等)等致命性毒性。
Hironobu等[10]基于日本人的研究表明,UGT1A1*6基因型虽未见于高加索人和非洲人群,但日本人发生率几乎等同于UGT1A1*28,因此,也应相应调整给药剂量。
约20%~30%患者接受以伊立替康为基础的联合化疗时发生3~4级迟发性腹泻和中性粒细胞减少,导致患者生活质量下降,甚至死亡。美国FDA2005年批准UGT1A1*28检测用于预测伊立替康不良反应,UGT1A1*28检测阳性患者应降低剂量(约30%),但仅适于给药剂量大与250 mg·m-2患者,不适于筛查国内患者与伊立替康低剂量治疗相关的不良反应。UGT1A1*6突变酶活性降低70%,与伊立替康60 mg·m-2以上剂量的不良反应相关,因此UGT1A1*6基因型个体,75 mg(30% 250 mg·m-2)恰为国人所应下调到的最低剂量。
2.3CYP3AP450 3A是参与药物氧化代谢的重要酶系,广泛存在于肝脏及肠道。人体参与药物代谢的CYP3A亚系为CYP3A3、CYP3A4、CYP3A5、CYP3A7。其中,CYP3A3被认为是CYP3A4的一种等位基因突变型,人体无活性表达;CYP3A7主要存在于胚胎肝脏;CYP3A4占肝脏及肠道CYPs总量的50%以上,参与约60%以上的药物催化代谢[13]。CYP3A5主要表达于肠壁、肾以及胰腺、前列腺和肺等肝外组织,不同于CYP3A4,与性别无关,且体内、体外不被外源性物质诱导。通过测定纯化表达的cDNA克隆,证明CYP3A5与CYP3A4具有相同底物结合特性,但活性比CYP3A4低80%[14]。
CYP3A基因位于人体第7号染色体q21.3~22.1。CYP3A的基因表达存在明显的个体差异,据人类细胞色素P450基因多态性命名委员会(Human Cytochrome P450 Allele Nomenclature Committee)数据,已发现24个CYP3A4单核苷酸基因突变体[15]。显然,CYP3A突变,导致其活性改变,导致许多常用药物效应改变。
CYP3A表达减少或活性受到抑制,伊立替康代谢产物APC及NPC生成减少,一定程度上增加SN-38浓度,从而增加发生SN-38浓度相关性不良反应的风险。相反,CYP3A活性增强,APC及NPC的生成增加,可以在一定程度上减小SN-38浓度。SAI等[16]研究发现,CYP3A4*1G携带者伊立替康血浓度-时间曲线下面积(AUC)上升20%。Maekawa等[17]对东亚人的研究发现,CYP3A4*16个体,表示伊立替康代谢为NPC速率的米氏常数(Km)显著高于CYP3A4*1(P<0.01),Vmax/Km较CYP3A4*1降低62%(P<0.05)。董宁宁等[18]对转移性结直肠癌患者氟尿嘧啶/亚叶酸钙/伊立替康(FOLFIRI)方案的研究表明,63例患者中,携带CYP3A5 GG基因型患者化疗有效率(完全缓解+部分缓解)显著高于携带AG/AA基因型患者(57.1% vs32.1%,P=0.048),Kaplan-Meier生存分析显示携带CYP3A5 GG基因型患者疾病进展时间显著长于AG/AA基因型患者(P=0.012)。
2.4β-葡萄糖醛酸酶(β-glucuronidase,β-G)β-G主要参与类固醇代谢,属溶酶体中水解酶类,广泛存在于人体组织及体液中,特别是肝、脾、肾上腺、肠黏膜及胃粘膜等,可人体自身合成,也可由多种肠道细菌产生。杨正时等[19]采用纸片法检测376株细菌产生β-G、半乳糖苷酶和色氨酸酶 (GTT试验)的能力,结果显示,97%的大肠杆菌,47.4%的志贺氏菌,41.1%的沙门氏菌能产生β-G。
由于无活性代谢产物SN-38G在肠道菌群β-G作用下可转化为SN-38,引起SN-38浓度增加,从而导致肠黏膜损害和肠上皮细胞脱落,杯状细胞和隐窝细胞不成比例增加和非典型性增生,破坏绒毛细胞的重吸收功能,导致肠腔内液体增加,最终导致小肠内吸收和分泌功能失去平衡,从而引起严重的腹泻。因此,减少肠道内β-G的产生或抑制其活性,可减轻腹泻的严重程度,减少腹泻的发生率。抗生素可通过杀灭肠道内细菌而减少β-G生成,从而达到止泻的目的[20]。但是,长期使用抗生素有可能导致细菌耐药或肠道菌群失调,从而导致伊立替康肠道吸收减少,引发二次腹泻。
如果能找到一种选择性β-G抑制剂,使既不杀死肠道细菌又可阻断SN-38G转化为SN-38,就可以较好地解决伊立替康引起的腹泻问题。Bret等[21]发现,细菌产生的β-G的分子结构与人类β-G有显著的差别,其包含的17个氨基酸形成的“微生物环”为人类β-G所不具有。以此作为β-G抑制剂的结合位点,通过高通量筛选,参考X-光衍射晶体结构,筛选近万个类似化合物,最后确定能选择性地抑制肠内细菌中β-葡萄糖醛酸酶活性而不影响肠内细菌正常生理功能4个先导化合物(图2)。

图2 选择性抑制肠内细菌β-葡萄糖醛酸酶活性而不影响 肠内细菌正常生理功能的先导化合物
2.5孕烷X受体(PXR)PXR又称类固醇和外源性检测核受体(steroid and xenobiotic sensing nuclear receptor,SXR)、孕烷激活受体(Pregnane-activated Receptor,PAR)等,是配体活化的转录因子超家族中的一员,系统名为核受体超家族1组Ⅰ成员2(NR1 Ⅰ 2),1998年由Kliewer等[22]首次克隆,因能被孕烷激活而得名;其下游效应靶基因均为主司异源性药物(毒物)及内源性激素代谢功能的酶及转运蛋白,其中包括CYP酶系在内的Ⅰ相代谢酶、葡萄糖醛酸转移酶等Ⅱ相代谢酶及ABC转运蛋白超家族的跨膜转运蛋白,如P-gp(P-糖蛋白)等药物转运蛋白,参与药物或毒物的体内处置,稳定机体内环境。PXR不仅在肝、结肠、肾、前列腺、乳腺、胃、心脏、外周血单核细胞、免疫细胞等正常组织中表达,还在结肠癌、乳腺癌、前列腺癌、子宫内膜癌、卵巢癌、骨肉瘤等肿瘤组织中表达,在小鼠、家兔和人类,PXR表达最丰富的器官是肝脏,其次是结肠和小肠。
PXR可显著影响CPT-11的敏感性。史祖宣等[23]研究表明,给予莱菔硫烷下调结肠癌细胞中PXR表达,结肠癌LS174T细胞对CTP-11化疗敏感性增加;给予利福平上调结肠癌细胞中PXR表达,LS174T细胞对CTP-11化疗敏感性减弱,且具有统计学意义。提示结肠癌细胞中PXR表达变化对于CPT-11化疗敏感性具有十分重要的影响,可能在结肠癌多药耐药及逆转多药耐药机制中具有重要作用。
2.6ABC转运蛋白ABC转运蛋白超家族是一组跨膜蛋白,包含100余种膜转运蛋白,广泛存在于细菌、植物和哺乳动物的各种细胞中,具有排出有毒物质、摄人营养物质、转运离子、多肽和细胞信号等功能。根据结构,ABC转运蛋白可分为全转运子和半转运子,全转运子含有2个ATP结合位点和2个疏水性结构区,半转运子含有1个ATP结合位点和1个疏水性结构区。ABC转运蛋白家族在跨膜区构成转运通道,可经由该通道转运许多结构不同物质穿过细胞膜。ABC转运蛋白超家族可分为多个亚家族(ABCA~ABCG),与肿瘤多药耐药相关的家族成员为ABCB、ABCC、ABCG等亚家族。
ABCB1即P-gp,由多药耐药基因1(multidrug resistance1,MDR1)编码,可通过其疏水位点与疏水性抗肿瘤药物结合,ATP水解供能,使逆浓度梯度将药物泵出细胞。高加索人的研究表明[24],ABCB1 1236C>T参与CPT-11转运,可使CPT-11和SN-38暴露量显著增加。但ABCA1*2单倍体基因型(包括ABCB1 1236C>T,ABCB1 2677G>A/T和ABCB1 3435C>T)日本患者,CPT-11、SN-38及APC肾清除率显著下降[25]。
ABCG2又名乳腺癌耐药蛋白(breast cancer resistance protein,BCRP),因其首先于乳腺癌细胞获得而得名。BCRP过度表达可使细胞内SN-38和SN-38G的浓度显著降低,从而显著降低SN-38的活性[26]。92例接受伊立替康治疗的结肠癌患者研究发现,伊立替康临床效应与ABCG2基因34G>A和376C>T位点单核苷酸多态性无关,但与421C>A位点单核苷酸多态性显著相关,其中C/C基因型的临床获益率最高[27]。
3展望
CPT-11或其活性代谢物SN-38,作为选择性拓扑异构酶抑制剂,并不阻碍拓扑异构酶与DNA结合,而是形成影响DNA功能的拓扑异构酶-抑制剂-DNA复合物,发挥杀灭瘤细胞作用。因此拓扑异构酶浓度越高,对抑制剂越敏感。多种肿瘤细胞,特别是结肠癌、卵巢癌、宫颈癌等细胞,尤其S期肿瘤细胞Topo Ⅰ含量远高于正常组织,从而对增殖期肿瘤细胞DNA复制有更强选择性抑制作用。
伊立替康临床应用广泛,致命性不良反应主要有迟发性腹泻或中性粒细胞减少,以及恶心呕吐、急性胆碱能综合征、肝肾功能损害、脱发等,与药物代谢酶、转运体遗传多态性有关,因地域、种族不同而异,代谢过程复杂,影响因素较多,其体内处置过程具有明显的个体差异,从而导致药效的个体差异。因此,进行基因检测,实施以基因检测结果为依据的个体化治疗,对于提高药效,减轻或防止不良反应的发生,提高患者生存质量有着积极的意义。
UGT1A1基因多态性机制探讨和临床应用研究,包括与基因突变有关的不同UGT1A1活性研究,不同基因型个体CPT-11及SN-38浓度与伊立替康效应相关性研究,伊立替康给药方案的制订、修饰、调整,以及SN-38新剂型开发研究,旨在实现以基因检测为依据的伊立替康个体化治疗。美国FDA要求在CPT-11药品标签上加入警示,建议使用CPT-11患者检测UGT1A1*28突变基因,同时为我国许多医疗机构、临床医生所接受,并取得满意成果。
参考文献:
[1] Hicks LD,Hyatt JL,Stoddard S,et al.Improved,Selective,Human Intestinal carboxylesterase inhibitors designed to nodulate 7-ethyl-10-[4-(1-piperidino) -1-piperidino]carbonyloxy-camptothecin (irinotecan;CPT-11) toxicity[J].J Med Chem,2009,52(12):3742-3752.
[2] Chabot GG,Abigerges D,Catimel G,et al.Population pharmacokinetics and pharmacodynamics of irinotecan (CPT-11) and active metabolite SN-38 during phase I trials[J].Ann Oncol,1995,6(2):141-151.
[3] Cersosimo RJ.Irinotecan:a new antineoplastic agent for the management of colorectal cancer[J].Ann Pharmacother,1998,32(12):1324-1333.
[4] Abrigerges D,Chabot GG,Armand JP,et al.Phase I and pharmacologic studies of the camptothecin analog irinotecan administered every 3 weeks in cancer patients[J].J Clin Oncol,1995,13(1):210-221.
[5] Humerickhouse R,Lohrbach K,Li L,et al.Characterization of CPT-11 hydrolysis by human liver carboxylesterase isoforms hCE-1 and hCE-2[J].Cancer Res,2000,60(5):1189 -1192.
[6] Kim SR,Sai K,Tanaka-Kagawa T,et al.Haplotypes and a novel defective allele of CES2 found in a Japanese population[J].Drug Metab Dispos,2007,35(10):1865-1872.
[7] Tanimoto K,Kaneyasu M,Shimokuni T,et al.Human carboxylesterase 1A2 expressed from carboxylesterase 1A1 and 1A2 genes is a potent predictor of CPT-11 cytotoxicity in vitro[J].Pharmacogenet Genomics,2007,17(1):1-10.
[8] Sai K,Saito Y,Tatewaki N,et al.Associat of carboxylesterase 1A genotypes with irinotecan pharmacokinetics in Japanese cancer patients[J].Br J Clin Pharmacol,2010,70 (2):222-233.
[9] 周喜.CES1基因多态性与伊立替康治疗晚期结直肠癌疗效的相关性研究[D].苏州:苏州大学,2012.
[10] Minami H,Sai K,Saeki M,et al.Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese:roles of UGT1A1*6 and *28[J].Pharmacogenet Genomics,2007,17(7):497-504.
[11] Carlini LE,Meropol NJ,Bever J,et al.UGT1A7 and UGT1A9 polymorphisms predict response and toxicity in colorectal cancer patients treated with capecitabine/irinotecan[J].Clin Cancer Res,2005,11(3):1226-1236.
[12] Paoluzzi L,Singh AS,Price DK,et al.Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38[J].J Clin Pharmacol,2004,44(8):854-860.
[13] Peyronneau MA,Renaud J P,Jaouen M,et al.Expression in yeast of three allelic cDNAs coding for human liver P4503A4 different stabilities,binding properties and catalytic activities of the yeast-produced enzymes[J].Eur J Biochem,1993,218:355-361.
[14] Jounaidi Y,Guzelian PS,Maurel P,et al.Sequence of the 5′-flanking region of CYP3A5:comparative analysis with CYP3A4 and CYP3A7[J].Biochem Biophys Res Commun,1994,205(3):1741-1747.
[15] Sim SC.CYP3A4 allele nomenclature[EB/OL].(2013-07-04)[2014-03-2].http://www.cypalleles.ki.se/cyp3a4.htm.
[16] Sai K,Saito YO,Fukushima-Uesaka H,et al.Impact of CYP3A4 haplotypes on irinotecan pharmacokinetics in Japanese cancer patients[J].Cancer Chemother Pharmacol,2008,62(3):529-537.
[17] Maekawa K,Harakawa N,Yoshimura T,et al.CYP3A4*16 and CYP3A4*18 alleles found in East Asians exhibit differential catalytic activities for seven CYP3A4 substrate drugs[J].Drug Metab Dispos,2010,38(12):2100-2104.
[18] 董宁宁,罗晓雅,何振.CYP3A5、GSTP1基因多态性与转移性结直肠癌患者化疗疗效的关系[J].临床和实验医学杂志,2014,13(8):607-610.
[19] 杨正时,钟熙,谢奉喻,等.通过葡萄糖醛酸酶-半乳糖苷酶-色氨酸酶(GGT)试验快速检定大肠杆菌的研究[J].微生物学通报,1989,16(2):88-92.
[20] Kehrer DF,Sparreboom A,Verweij J,et al.Modulation of irinotecan-induced diarrhea by cotreatment with neomycin in cancer patients[J].Clin Cancer Res.2001,7(5):1136-1141.
[21] Wallace BD,Wang H,Lane KT,et al.Alleviating cancer drug toxicity by inhibiting a bacterial enzyme[J].Science,2010,330(6005):831-835.
[22] Kliewer SA,Moore JT,Wade L,et al.An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway[J].Cell,1998,92(1):73-82.
[23] 史祖宣,汤喻,李克,等.结肠癌细胞核受体PXR表达对CPT-11化疗效果的影响[J].重庆医科大学学报,2011,36(11):1381-1383.
[24] Mathijssen RH,Marsh S,Karlsson MO,et al.Irinotecan pathway genotype analysis to predict pharmacokinetics[J].Clin Cancer Res,2003,9(9):3246-3253.
[25] Sai K,Kaniwa N,Itoda M,et al.Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan[J].Pharmacogenetics.2003,13(12):741-757.
[26] Kawabata S,Oka M,Shiozawa K,et al.Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells[J].Biochem Biophys Res Commun.2001,280(5):1216-1223.
[27] 吴慧娟,吴红波,邹宏志,等.结肠癌中ABCG2基因多态性与伊立替康疗效的相关性研究[J].中国现代医学杂志,2014,24(7):48-50.
猜你喜欢
渔业科学进展(2022年4期)2022-09-05
世界科学技术-中医药现代化(2022年3期)2022-08-22
承德医学院学报(2022年2期)2022-05-23
中国典型病例大全(2022年9期)2022-04-19
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2022年2期)2022-03-29
临床肝胆病杂志(2021年3期)2021-12-03
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年5期)2021-07-22
昆明医科大学学报(2021年2期)2021-03-29
