
一个遗传性凝血因子Ⅺ缺陷症家系的基因分析
2015-12-27 22:12:08戴利亚张德亭谢海啸张扬郑芳秀王明山温州市人民医院检验科浙江温州35000温州医科大学附属第一医院检验科浙江温州3505
温州医科大学学报 2015年5期
戴利亚,张德亭,谢海啸,张扬,郑芳秀,王明山(.温州市人民医院 检验科,浙江 温州 35000;.温州医科大学附属第一医院 检验科,浙江 温州 3505)
·临 床 经 验·
一个遗传性凝血因子Ⅺ缺陷症家系的基因分析
戴利亚1,张德亭1,谢海啸2,张扬2,郑芳秀2,王明山2
(1.温州市人民医院 检验科,浙江 温州 325000;2.温州医科大学附属第一医院 检验科,浙江 温州 325015)
目的:对一个遗传性凝血因子Ⅺ(FⅪ)缺陷症家系进行FⅪ基因突变的分析和家系调查,探讨其分子发病机制。方法:检测先证者及其家系成员活化部分凝血活酶时间(APTT)、凝血酶原时间(PT)、凝血因子Ⅷ活性(FⅧ:C)、凝血因子Ⅸ活性(FⅨ:C)、凝血因子Ⅻ活性(FⅫ:C)、FⅪ活性(FⅪ:C)和FⅪ抗原(FⅪ:Ag)含量等进行表型诊断;用DNA直接测序法分析先证者FⅪ基因所有15个外显子、侧翼、5’和3’非翻译区及家系成员相应的突变位点区域,用反向测序证实所发生的突变。结果:先证者APTT延长为50.0 s (正常为27.0~41.0 s),先证者弟弟与儿子APTT同样延长,分别为53.7 s和51.8 s,家系其他成员APTT无明显延长;先证者及其弟弟、儿子、父亲的FⅪ:C明显减低,分别为22.5%、22.3%、35.5%和42.0%,FⅪ:Ag含量分别为29.0%、18.0%、29.0%和40.0%,表现为交叉反应物质(CRM)阴性;先证者母亲凝血指标均在参考范围内。基因测序发现先证者及其父亲、弟弟、儿子FⅪ基因第8号外显子存在C16642T杂合无义突变,导致编码第263位氨基酸谷氨酰胺提前出现终止密码(Gln263stop)。结论:FⅪ基因第8号外显子C16642T杂合无义突变是导致该遗传性FⅪ缺陷症家系FⅪ水平降低的主要原因。
凝血因子Ⅺ;家系;杂合;无义突变
遗传性凝血因子XI(FXI)缺陷症为常染色体隐性遗传性疾病,曾被称为血友病C,患者出血症状轻重不定,FXI水平高低与出血程度并不成正比,很少有自发性出血[1]。近年来的报道认为,该缺陷症的发生主要与FXI基因的突变有关[2-3]。本研究对一个遗传性FXI缺陷症家系进行FXI基因突变分析和家系调查,初步探讨其分子发病机制。
1 对象和方法
1.1 对象
1.1.1 家系资料:该家系共3代5名成员,包括先证者及其父亲、母亲、弟弟和儿子。先证者,女,20岁,因“妊娠待产”收住入院。实验室检查:活化部分凝血活酶时间(APTT)延长为50.0 s;凝血因子检查发现FXI活性(FXI:C)为22.5%,FXI抗原含量(FXI:Ag)为29.0%。先证者肝肾功能正常,平素无明显出血症状,无长期服用药物史。先证者父母非近亲结婚,询问其家系成员均无出血史,家系谱见图1。
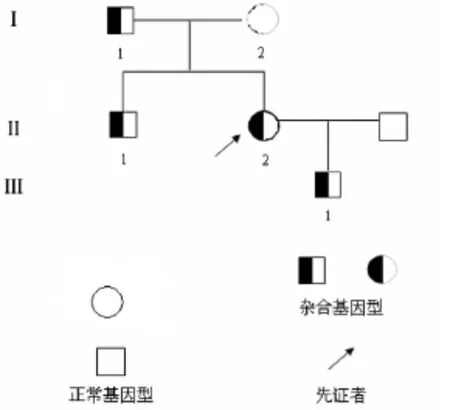
图1 遗传性FXI缺陷症家系图
1.1.2 正常对照组:共106名,男62名,女44名,平均年龄32岁(16~60岁)。无肝肾功能疾病,无血栓出血史。征求本人同意,对上述人员采集血样。
1.2 仪器与试剂
1.2.1 仪器:STA-R全自动血凝仪(法国STAGO公司)、电泳仪(Bio-rad公司)、PCR扩增仪(ABI Thermal cycler 2720)、凝胶成像系统(上海天能公司)。
1.2.2 引物:根据NCBI中的FXI基因序列(Genbank AY191837),应用Primer 5软件共设计13对引物以覆盖FXI基因的15个外显子区域及其侧翼序列,由上海桑尼生物科技有限公司合成,见表1。
1.2.3 试剂:APTT、FXI:C等凝血试剂由法国STAGO公司配套提供;PCR试剂盒(Master Mix)由北京天根生化科技有限公司提供。
1.3 方法
1.3.1 标本采集和处理:采集实验对象外周血标本2管,0.109 mol/L枸橼酸钠1:9抗凝,一管用于凝血功能检测;另一管用于抽提DNA,-40 ℃冻存,用于PCR扩增。

表1 FXI各引物序列及其PCR实验的退火温度
1.3.2 凝血功能检测:APTT、凝血酶原时间(PT)、凝血因子VIII活性(FVIII:C)、凝血因子IX活性(FXI: C)、凝血因子XII活性(FXII:C)均采用一期凝固法;FXI:Ag采用酶联免疫吸附(ELISA)法检测。
1.3.3 DNA抽提:使用北京天根生化科技有限公司基因组DNA提取试剂盒抽提先证者及家系成员外周血基因组DNA。
1.3.4 PCR扩增:反应体系:反应体积共50 μL,包括Taq PCR Mastermix 25μL,ddH2O 18μL,DNA模板3μL,正向引物2μL,反向引物2μL。反应条件:95 ℃预变性5 min,95 ℃变性30 s,根据不同引物分别以相应退火温度(53~60 ℃)退火30 s,72 ℃延伸45 s(共35个循环),72 ℃延伸10 min。
1.3.5 电泳:5μL PCR产物采用Goldview标记,在 1.5%琼脂糖凝胶上进行电泳。
1.3.6 DNA序列分析:①家系分析:PCR扩增产物送上海桑尼生物工程有限公司测序,所用测序仪为ABI PRISM 3730。测序结果用Chromas软件与美国NCBI基因库所公布的FXI基因序列(Genbank AY191837) 进行比对,寻找基因突变位点。发现基因突变的序列则反向测序予以证实。②正常对照:对正常对照组人群相应的区域进行PCR扩增、测序,排除基因多态性。
2 结果
2.1 先证者及家系成员凝血功能检测结果 先证者APTT延长为50.0 s,先证者弟弟与儿子APTT同样延长,分别为53.7 s和51.8 s,家系其他成员APTT无明显延长;先证者及其父亲、儿子、弟弟的FXI: C和FXI:Ag均有不同程度下降,结果见表2。

表2 FXI缺陷症家系表型检测结果
2.2 FXI基因分析结果 先证者及其父亲、弟弟、儿子FXI基因第8号外显子存在C16642T杂合无义突变,导致编码第263位氨基酸谷氨酰胺提前出现终止密码(Gln263stop);对106名健康对照进行筛查,排除了基因多态性的可能。基因型检测结果见表3、图2-3。

表3 先证者及家庭成员基因检测结果

图2 先证者FXI基因第8号外显子的测序结果(16642位C>T导致Gln263stop 杂合突变)
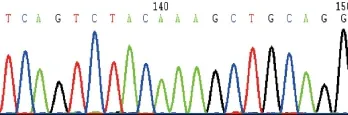
图3 先证者母亲FXI基因第8号外显子相应位置测序图(正常野生型)
3 讨论
FXI的编码基因位于4号染色体长臂(4q35),总的基因长度为23 kb,含有15个外显子和14个内含子[4]。成熟的FXI在肝细胞和巨核细胞中合成,由2条相同分子量的多肽链通过二硫键连接形成二聚体。有报道[5]指出该同源二聚体结构对FXI的有效分泌与功能发挥起着重要作用。FXI显著的结构特征为4个AP结构域形成盘状结构及其所连接的催化域,4个AP结构域有以下功能:AP1上有凝血酶结合位点;AP2上有HMWK结合位点;AP3与肝素、FIX、血小板膜糖蛋白(GP)Ib的结合有关;AP4第321位的半胱氨酸(Cys321)是2个FXI单体间通过二硫键形成 二聚体的部位[6]。
本资料先证者因“妊娠待产”行常规凝血功能检测,发现APTT明显延长,FXI:C和FXI:Ag含量明显降低,因其抗原与活性水平均同步下降,属于CRM阴性型。而其他内源凝血途经的凝血因子活性及抗原含量均在参考范围之内,临床上诊断其为FXI缺陷症。随后的基因测序分析结果证明,先证者FXI基因第8号外显子存在C16642→T杂合无义突变,从而导致编码的263号氨基酸谷氨酰胺发生无义突变。家系分析发现先证者父亲、弟弟及儿子存在同样的突变位点,其FXI:C和FXI:Ag含量均有不同程度下降,表明先证者和其弟弟FXI基因第8号外显子突变均遗传于父亲,而后又遗传给儿子,因此可诊断为遗传性FXI缺陷症。王静等[4]在1例遗传性FXI缺陷症家系基因缺陷研究中同样发现了先证者FXI基因第8号外显子存在C16642→T杂合无义突变,不同的是,该先证者FXI基因第2号外显子还存在G3733C错义突变。分析认为E2区的错义突变导致编码最后一位信号肽的氨基酸发生G>R替换,从而直接影响内质网上信号肽的剪切,使得FXI蛋白质到内质网腔分泌障碍;而E8的无义突变使得编码263位的氨基酸提前出现终止密码,形成截短型蛋白,该蛋白因缺失部分结构域,高度不稳定,并伴细胞内迅速降解。笔者分析该双重杂合突变导致其FXI:C及FXI:Ag明显下降。而在本研究中,家系成员除先证者母亲正常外,其余均为单一位点突变杂合子,因此认为FXI基因第8号外显子C16642T杂合无义突变是导致该遗传性FXI缺陷症家系FXI水平降低的主要原因。Quélin等[7]指出遗传性FXI缺陷症中基因突变的纯合子和双重突变杂合子可致血浆FXI:C下降至15%以下,单一位点突变杂合子为15%~70%。本研究中有基因突变的家系成员FXI:C水平与此相符。此外在1个中国香港人及2个日本人家系研究中发现的情况同样如此,且对于该突变在东方人群中的一定流行率起一定支持作用[8-9],但仍有待更多的数据进一步证明。
FXI的第7、第8号外显子编码AP3结构域,因此任何该二区的突变都可能会干扰FXI蛋白中AP3结构域的结构形成从而影响相关的功能。随着当前FXI晶体结构的不断完善,AP3结构域的功能也不断被发现,有结合FIX、肝素及血小板GPIb的作用。根据传统的内源性凝血途径,由FXIa激活FIX来维持凝血过程继续进行,FXI通过AP3结构域结合FIX,催化裂解FIX的Arg145-Ala146键,而后裂解Arg180-Val181键,最终活化FIX[10]。尽管在接触系统中,血小板并不能促进FXI的活化,然而更多的证据显示在流体状态下血小板能影响FXI及FXIa行为。虽然FXI缺乏γ-羟基谷氨酸结构域,但强有力的证据显示FXI可结合血小板[11-12]。Greengard等[13]认为活化的血小板上约有1 500个FXI结合位点,由其上的血小板GPIb介导连接。另有报道[14]认为FXI与血小板结合的最佳条件为FXI上的结合位点、血小板GPIb的氨基末端结构域及HMWK和锌离子,进一步支持上述论断。研究报道描述凝血酶首先利用其前表面阴离子结合位点I与FXI AP1结构域结合,再利用其后表面阴离子结合位点II与GPIb-IX-V复合物结合,所以当FXI一单体的AP3结构域与GPIb结合时,可通过另一单体的A1结构域与相邻的凝血酶反应,从而得到激活。新的研究认为FXI结合在血小板受体上,一种称之为载脂蛋白E的物质,血小板上该受体的数量 与FXI结合位点数量相符[12]。有报道[15]指出,FXI AP3结构域中Ser248、Arg250、Lys255、Phe250及 Gln263为血小板的结合位点。本实验中所检测到的突变位点即为Gln263Stop。除却王静等[4]认为无义突变发生时,mRNA水平可正常,但生成截短形式的蛋白质高度不稳定,并迅速被降解,也有研究[16]认为无义突变产生的含有提前终止密码子的mRNA可被无义介导衰变的mRNA监视系统迅速降解。前者被认为是循环中FXI减少的主要原因。
FXI基因缺陷发病相关的分子机制尚不完全清楚,即使有报道利用计算机软件来对正常与异常FXI空间结构进行比较,从而利于了解基因缺陷对蛋白空间构型变化和功能的影响,但仍缺少相关的客观依据。因此本研究对于今后建立体外表达系统,以进一步研究该因子基因突变致病的分子机制起一定的指导作用。
[1] Meijiers JC,Tekelenburg WL,Bouma BN,et al.High levels of coagulation factor XI as a risk factor for venous thrombo-sis[J] .N Engl J Med,2000,342(10): 696-701.
[2] Solda G,Asselta R,Ghiotto R,et al.A type II mutation (Glu117Stop) induction of allele-specific mRNA degradation and factor XI deficiency[J] .Haematologica,2005,90(12): 1716-1718.
[3] Kravtsov DV,Monahan PE,Gailani D.A classification sys-tem for cross-reactive material-negative factor XI deficiency [J] .Blood,2005,105(12): 4671-4673.
[4] 王静,傅启华,王学锋,等.遗传性凝血因子Ⅺ缺陷症一例(家系基因缺陷研究)[J] .中华检验医学杂志,2009,32(7): 794-797.
[5] Wu W,Sinha D,Shikov S,et al.Factor XI homodimer struc- ture is essential for normal proteolytic activation by factor XIIa,thrombin and factor XIa[J] .J Biol Chem,2008,283 (27): 18655-18664.
[6] Gailani D,Smith SB.Structural and functional features of factor XI[J] .J Thromb Haemost,2009,7(suppl 1): 75-78.
[7] Quélin F,Mathonnet F,Potentini-Fsault C,et al.Identifica-tion of five novel mutaitons in the factor XI gene (F11) of patients with factor XI deficiency[J] .Blood Coagul Fibrino-lysis,2006,17(1): 69-73.
[8] Au WY,Cheung JW,Lam CC,et al.Two factor XI muta-tions in a Chinese family with factor XI deficiency[J] .Am J Hematol,2003,74(2): 136-138.
[9] Sato E,Kawamata N,Kato A,et al.A novel mutation that leads to a congenital factor XI deficiency in a Japanese fam-ily[J] .Am J Hematol,2000,63(14): 165-169.
[10] Lawson Jh,Mann KG.Cooperative activation of human fac-tor IX by the human extrinsic pathway of blood coagulation [J] .J Biol Chem,1991,266(17): 11317-11327.
[11] Tucker EI,Marzec UM,White TC,et al.Prevention of vas-cular graft occlusion and thrombus-associated thrombin gen-eration by the inhibiton of fator XI[J] .Blood,2009,113(4): 936-944.
[12] White-Adams TC,Berny MA,Tucker EI,et al.Identifica-tion of coagulation factor XI as a ligand for platelet apolipo-protein E receptor 2 (ApoER2)[J] .Arterioscler Thromb Vasc Biol,2009,29(10): 1602-1607.
[13] Greengard JS,Heeb MJ,Ersdal E,et al.Binding of coagu-lation factor XI to washed human platelets[J] .Biochemistry,1986,25(13): 3884-3890.
[14] Baglia FA,Gailani D,Lopez JA,et al.Identification of a binding site for glycoprotein Ibα in the apple 3 domain of factor XI[J] .J Biol Chem,2004,279(44): 45470-45476.
[15] Sun MF,Baglia FA,Ho D,et al.Defective binding of factor XI-N248 to activated human platelets[J] .Blood,2001,98 (1): 125-129.
[16] Iijima K,Udagawa A,Kawasaki H,et al.A factor XI defi-ciency associated with a nonsense mutation (Trp501stop) in the catalytic domain[J] .Br J Haetmatol,2000,111: 556-558.
(本文编辑:丁敏娇)
The gene analysis of a Chinese pedigree with congenital blood coagulation factor XI deficiency
DAI Liya1,ZHANG Deting1,XIE Haixiao2,ZHANG Yang2,ZHENG Fangxiu2,WANG Mingshan2.
1.Department of Laboratory,Wenzhou People’s Hospital,Wenzhou,325000; 2.Department of Laboratory,the First Affiliated Hospital of Wenzhou Medical University,Wenzhou,325015
Objective:To analyze genetic mutation and explore its molecular pathogenesis for a Chinese pedigree with congenital blood coagulation factor XI deficiency.Methods:Activated partial thromboplastin time (APTT),Prothrombin time (PT),FXI activity (FXI:C),FXI antigen (FXI:Ag) and other coagulant parameters were assayed.Exons 1-15,exon-intron boundaries and 5’,3’ untranslated sequences of FXI gene of the proband and other family members were analyzed by direct sequencing.The detected mutations were confirmed by sequencing the complementary strand.Results:The proband had prolonged APTT (50.0 s).The APTT results of her brother and son were prolonged too,which were 53.7 second and 51.8 second respectively.Other members’APTT were in normal range.The activity and antigen of proband’s FXI was 22.5% and 29.0%,while her brotherwas 22.3% and 18.0%,her son was 35.5% and 29.0%,her father was 42.0% and 40% separately,which were called cross-reactivity material negative.The mother of proband had normal results above-mentioned.After theamplification and sequencing of 15 extrons in FXI,an exon 8 C16642→T mutation in proband as well as in her father,brother and son was found,which led to an amino acid change of Gln→Term at residue 263.Conclusion:The mutation of C16642→T on exon 8 is the main reason for the congenital FXI deficiency in this Chinese pedigree.
blood coagulation factor XI; pedigree; heterozygosis; nonsense mutation
R394.3
B
10.3969/j.issn.2095-9400.2015.05.014
2014-07-21
戴利亚(1982-),女,浙江瑞安人,主管检验师。
王明山,副主任技师,硕士生导师,Email:wyms@ 126.com。
猜你喜欢
青年文学家(2024年10期)2024-05-26 18:27:59
北京大学学报(医学版)(2022年2期)2022-11-21 03:29:12
中国现代医生(2022年19期)2022-11-04 10:13:29
临床输血与检验(2022年3期)2022-06-22 02:52:50
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
遗传(2019年5期)2019-05-21 09:58:28
传染病信息(2019年2期)2019-05-17 13:16:04
