超高效液相色谱-四极杆飞行时间质谱法快速筛查水产品中15 种碱性合成色素
2015-12-26 01:59:24戴意飞王萍亚赵巧灵蒋玲波罗海军
色谱 2015年10期
黄 鹂, 戴意飞, 王萍亚, 周 勇, 赵巧灵 , 蒋玲波, 罗海军
(舟山市食品药品检验检测研究院,舟山市质量技术监督检测研究院,浙江 舟山316021)
近年来,违法使用碱性橙、罗丹明B、孔雀石绿等工业碱性合成色素的食品安全问题尤为突出。这些工业碱性色素具有较强的致毒、致癌作用,由于色彩丰富、价格低廉、着色稳定,被某些不法分子非法添加在食品中,对人民健康和社会稳定造成危害,成为食品安全领域内的高危风险因子。因此,目前关于食品中碱性橙、罗丹明B、孔雀石绿等碱性色素的检测方法越来越受关注,已有较多报道[1-7]。
近几年有关色素的检测方法层出不穷,主要有分光光度法[8]、薄层色谱法[9]、毛细管电泳法[10]、示波极谱法[11]、酶联免疫吸附分析法[12]、高效液相色谱法[13-16]、液相色谱-串联质谱法[17,18]等,由于不同色素的极性不同,导致现有的检测方法在高灵敏度和高效方面具有一定的局限性。
四极杆飞行时间质谱(Q-TOF MS)能提供精确质量数,可通过分子质量数的匹配对化合物进行鉴定和确证,现在已开展了部分研究,如奶酪中色素的分析[19]、饮料中色素的快速检测[20]。但是目前使用Q-TOF MS 对于食品中未知添加物的筛查研究总体上比较少,尤其是对水产品中的色素筛查未查到相关报道。因此,本文以水产品为研究对象,对其中15 种禁用的碱性合成色素进行分析,并对样品前处理方法、液相色谱条件、质谱条件进行优化,以期建立水产品中多种色素的UPLC-Q-TOF MS 快速高效的检测方法。
1 实验部分
1.1 仪器与试剂
Agilent UHPLC 和Agilent 6540-QTOF(Agilent公司,美国);Avanti®J-E 高速冷冻离心机、Talboys 涡旋混合器、N-EVAPTM-112 氮吹仪、样品萃取管;甲醇、乙腈、甲酸、乙酸为色谱纯,均购自上海安谱实验科技股份有限公司;无水硫酸镁、乙酸铵为分析纯;C18填料(40~63 μm)、硅胶粉(40~63 μm)均购自上海安谱实验科技股份有限公司;阳离子交换小柱(Oasis MCX)、弱阳离子交换小柱(Oasis WCX)均购自Waters 公司;C18小柱购自上海安谱实验科技股份有限公司;实验用水为Milli-Q 超纯水。
标准品:金胺(Auramine O,纯度89%)、隐性孔雀石绿(Leucomalachite Green,纯度90%)、孔雀石绿(Malachite Green,纯度89%)、苏丹红III(Sudan III,纯度91%)、苏丹红IV (Sudan IV,纯度91%)、苏丹黄(Butter Yellow,纯度89%)、苏丹黑B(Sudan Black B,纯度91%)、碱性橙II(Chrysoidine G,纯度89%)、罗丹明B(Rhodamine B,纯度91%)、苏丹橙(Sudan Orange G,纯度89%)、苏丹红7B (Sudan Red 7B,纯度91%)、苏丹红G(Sudan Red G,纯度91%)、苏丹红I(Sudan I,纯度91%)、苏丹红II(Sudan II,纯度91%)、橘红2 号(Citrus Red 2,纯度91%),均购于上海安谱实验科技股份有限公司。
1.2 标准溶液配制
分别准确称取15 种碱性合成色素标准品各10 mg 于10 mL 容量瓶中,用甲醇溶解,配制成1 g/L的单标准储备液(-20 ℃保存),再分别吸取1 g/L的15 种单标准储备液各1 mL 于100 mL 容量瓶中,用甲醇定容得10 mg/L 的混合标准溶液。实验中使用初始流动相溶液将其稀释成相应的标准工作溶液,于4 ℃保存。
1.3 样品前处理
称取2.0 g 样品于50 mL 带盖聚四氟乙烯离心管中,加入10 mL 乙腈(含有1%(v/v)乙酸)溶液,涡旋1 min,超声5 min,以10 000 r/min 冷冻离心10 min,取5 mL 上清液转移至基质分散管中,管内装有C18复合硅胶吸附剂(500 mg 无水硫酸镁、100 mg C18粉末、40 mg 硅胶粉末),振荡基质分散管5 min,以10 000 r/min 冷冻离心10 min,取离心后的上清液3 mL,经氮气吹干,用甲醇-水(50 ∶50,v/v)溶解并定容到1 mL,过0.22 μm 有机滤膜后供UPLC-Q-TOF MS 分析测定。
1.4 色谱-质谱条件
1.4.1 色谱条件
Agilent Eclipse Plus-C18(100 mm×3.0 mm,1.8 μm)作为分析柱,柱温为30 ℃;流动相:A 相为5 mmol/L 乙酸铵-0.1%(v/v)甲酸水溶液,B 相为乙腈;梯度洗脱程序:0~1 min,5%B~20%B;1~10 min,20%B ~100%B;10 ~15 min,100%B。进样体积为3 μL,流速为0.2 mL/min。
1.4.2 质谱条件
离子源:电喷雾离子(ESI)源;扫描方式:正离子全扫描;全扫描范围:m/z 50 ~1 100;毛细管电压:4 000 V;离子源喷雾电压:250 V;离子化电压:130 V;鞘气温度:350 ℃;干燥气温度:325 ℃;鞘气流速:11 L/min;干燥气流速:8 L/min;数据采集模式:全息离子扫描(All ions MS/MS)。参比溶液中含三氟乙酸(C2HF3O2,其精确相对离子质量为112.985 5)和六(1H,1H,3H-全氟丙氧基)磷氮的加氢离子(C18H19F24N3O6P+3,其精确相对离子质量为922.009 8)。
数据采集与处理通过Agilent Mass Hunter Workstation Software (Version B.05.00)软件完成,碎片离子数据库建立通过Agilent Mass Hunter PCDL Manager (B.04.00)软件完成。
2 结果与讨论
2.1 色谱-质谱分析
本文以隐性孔雀石绿、苏丹红III 等为代表的15 种在水产品中禁用的碱性合成色素为研究目标物进行检测,依据它们的相对分子质量和理化性质的差异,结合应用UPLC 技术进行色谱分离,Q-TOF MS 进行质谱信息采集。图1 是15 种碱性合成色素的分子特征提取色谱图,并由All ions MS/MS 模式收集15 种目标物的母离子及保留时间等信息(见表1)。通过实验获得的质谱信息并结合Agilent Mass Hunter 软件对目标物进行质谱库对比,以保留时间、母离子和碎片离子精确质量数、同位素丰度模型进行比对确证,以母离子用于定量分析。由于方法中的碎片匹配采用了二级全离子图谱匹配,所以匹配度比一般用特征子离子的方式有更高的可靠性和准确度,效果更好,使得筛查的准确度更高。

图1 15 种碱性合成色素的UPLC-Q-TOF MS 提取离子色谱图Fig.1 Extracted chromatograms of the 15 basic artificial dyes by UPLC-Q-TOF MS

表1 水产品中15 种碱性合成色素的保留时间和质谱信息Table 1 Retention times and MS information for the 15 basic artificial dyes in fishery products
2.2 前处理条件的优化
2.2.1 提取溶剂的选择
提取溶剂的优化需同时考察目标化合物和样品基质的特性,合适的提取剂可有效分离目标物与基质杂质,从而增加对样品中目标物的检测灵敏度。本实验比较了4 种有机溶剂——乙腈、乙酸乙酯、二氯甲烷和丙酮,研究分析表明,乙腈最适合作为水产品中碱性合成色素的提取剂,回收率最高,达到90%。乙腈适用范围广,它不仅减少了对基质中油脂的溶解,且可有效沉淀蛋白质,减少杂质对色素的干扰。二氯甲烷因溶剂密度大,提取液分布于底层,增加操作难度,回收率为82%;此外,以丙酮和乙酸乙酯提取色素时,回收率偏低,均在60% 以下。在确定了最优提取剂的基础上,本文进一步分析了加入1%(v/v)乙酸的乙腈溶液对色素的提取效率,结果显示加入乙酸后提取剂不仅保持了较好的回收率(达到96%),且能使目标物离子化,为下一步提取液的分离净化处理奠定了良好基础。因此,本文确定最优提取溶剂是含1%(v/v)乙酸的乙腈溶液。
2.2.2 净化方法的优化
本实验选用3 种常用的固相萃取柱(C18柱、Oasis WCX 柱、Oasis MCX 柱)和C18复合硅胶基质萃取管对水产品中色素的提取分离效率进行比较分析,样品中色素的回收率结果见表2。由表2 可知,以C18复合硅胶基质萃取管为净化载体时,样品中色素的回收率最高,分析原因可能有以下几个方面:①C18固相萃取小柱具有强疏水性,但吸附性较弱,导致15 种碱性合成色素回收率较低(在60%~75%之间);②弱阳离子交换柱和阳离子固相萃取小柱平均色素回收率为85.13%和74.09%,但对个别目标物如碱性橙II、苏丹红II 等的吸附能力较弱,达不到检测方法要求的高通量检测要求;③从表2 可以看出,自制C18复合硅胶混合基质萃取管对15 种碱性合成色素的平均回收率高达96.58%,它通过不同吸附剂对目标物进行针对性的保留,有效地去除杂质干扰,提高了碱性合成色素的净化效率,缩短了检测周期。同时对于萃取管中吸附剂的量进行了考察,结果发现:无水硫酸镁的量在500 mg 及以上时处于过饱和状态,达到良好的盐析效果;当C18和硅胶的配比为100 mg ∶40 mg 及以上时,对于回收率和净化效果不再有明显的促进效果,因此考虑到成本及效果,最终选择了吸附剂的使用量为500 mg无水硫酸镁、100 mg C18粉末、40 mg 硅胶粉末。因此,本文确定使用C18复合硅胶基质萃取管作为净化载体,从而使15 种碱性合成色素物质有较好的回收率,为水产品中色素的检测方法提供了一个快速有效的净化手段。
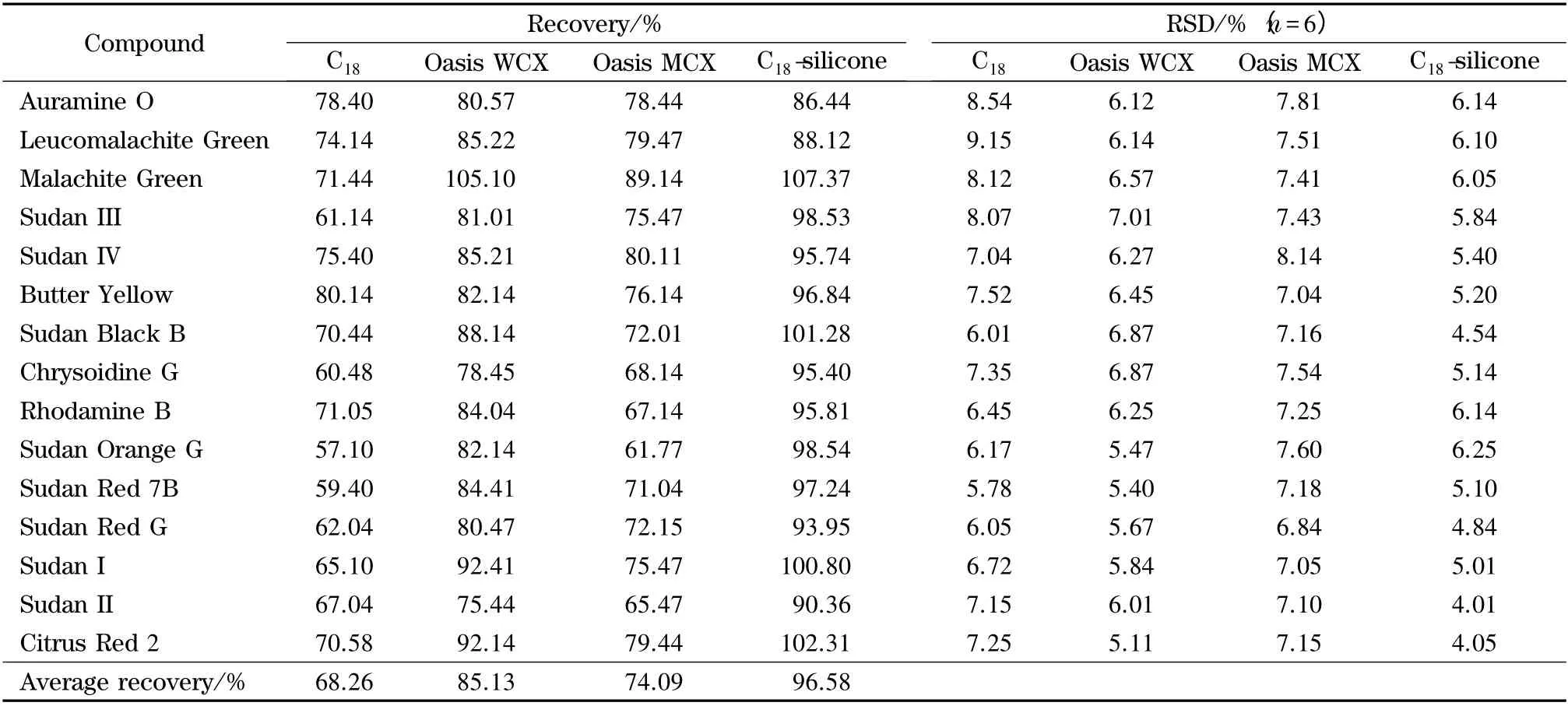
表2 不同萃取柱对水产品中15 种碱性合成色素的加标回收率及相对标准偏差的影响(n=6)Table 2 Influence on the spiked recoveries and RSDs of the 15 target compounds in fishery products by different SPE cartridges (n=6)
2.3 方法学确证
2.3.1 标准曲线、检出限与精密度
配制0.1 ~500 μg/L(0.1、0.2、0.5、1、2、5、10、20、50、100、200、500 μg/L)的混合标准溶液,以分子离子峰面积(Y)对相应的质量浓度(X,μg/L)分别绘制15 种碱性合成色素的标准曲线,并将10 μg/L 混合标准溶液平行测定6 次,计算峰面积的相对标准偏差(RSD),结果见表3。从表3 可知,15 种碱性合成色素在各自的线性范围内线性关系良好,相关系数(r)≥0.993;RSD≤6.49%,说明仪器精密度良好。
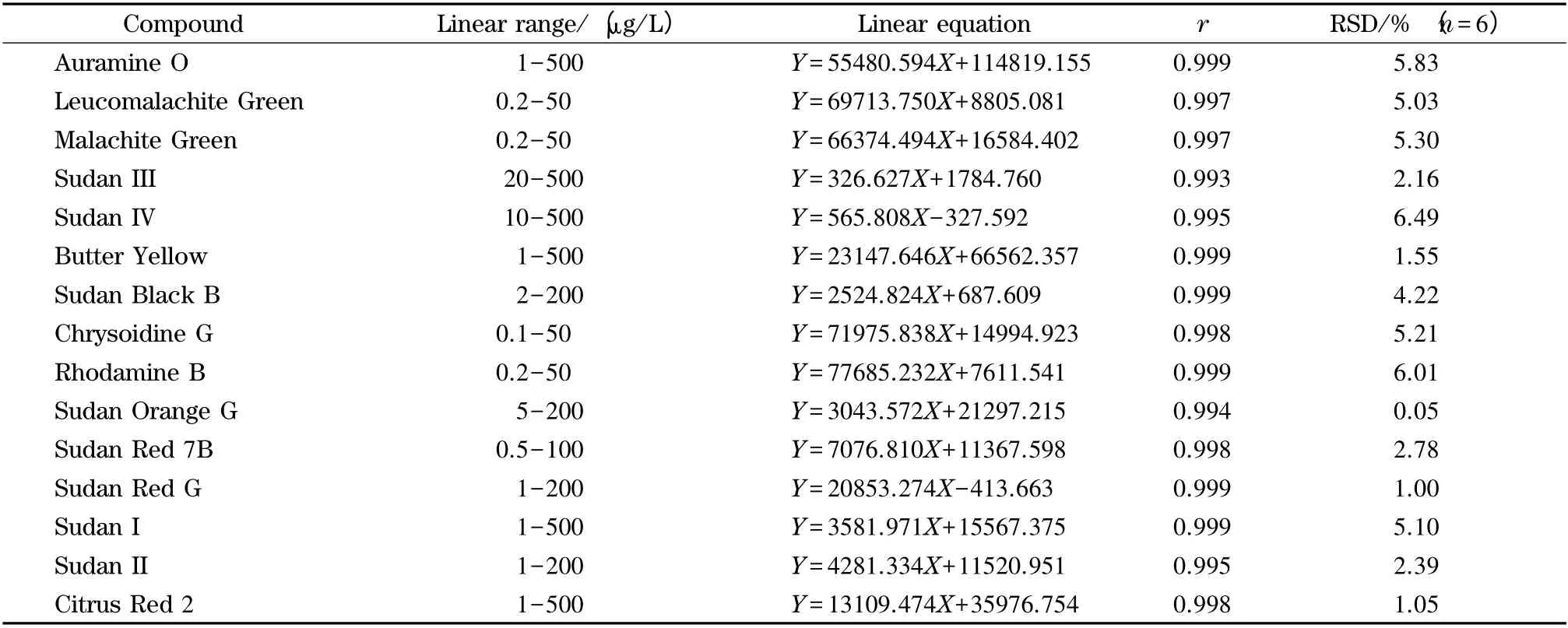
表3 15 种碱性色素的线性范围、线性方程、相关系数(r)和10 μg/L 时峰面积的相对标准偏差(RSD)Table 3 Linear ranges,linear equations,correlation coefficients (r)and relative standard deviations (RSDs)of the 15 basic artificial dyes at 10 μg/L
2.3.2 检出限、定量限与回收率
本文中的检出限与定量限是根据3 倍和10 倍信噪比的要求进行测定和计算的。实际操作中,我们根据10.0 μg/kg 的添加回收试验(苏丹红III 和苏丹红IV 的加标量分别为100.0 μg/kg 和50.0 μg/kg)中目标化合物检测到的色谱峰进行3 倍信噪比的换算,最后确定为检出限(LOD,S/N =3),而该色谱峰经10 倍信噪比换算得到的含量为定量限(LOQ,S/N =10),最终通过采用初始流动相来稀释,直到信噪比分别达到3 和10 时的含量,进行最终各目标物的检出限和定量限的验证。如表4 所示,15 种目标物的LOD 在0.05 ~2.0 μg/kg 之间(苏丹红III 和苏丹红IV 的检出限分别为50.0 μg/kg 和25.0 μg/kg),同时15 种色素的定量限小于5.0 μg/kg(苏丹红III 和苏丹红IV 的定量限分别为100.0 μg/kg 和50.0 μg/kg)。
在阴性的鲳鱼样品基质中添加10、50 和200 μg/kg 3 种不同浓度水平的15 种混合标准溶液(苏丹红III 和苏丹红IV 的添加浓度分别为100、500、20 000 μg/kg),按1.3 节方法进行预处理,采用UPLC-Q-TOF MS 测定提取液中的目标化合物,且每个水平重复测定6 次,计算平均回收率及RSD,结果见表4。由表4 可知,在不同加标浓度范围内,15 种碱性合成色素的回收率为80.60%~107.37%,RSD 为3.33%~6.69%。由15 种碱性合成色素的检出限、定量限和回收率的实验结果,可以看出本方法满足快速筛查水产品中15 种碱性合成色素的需要。
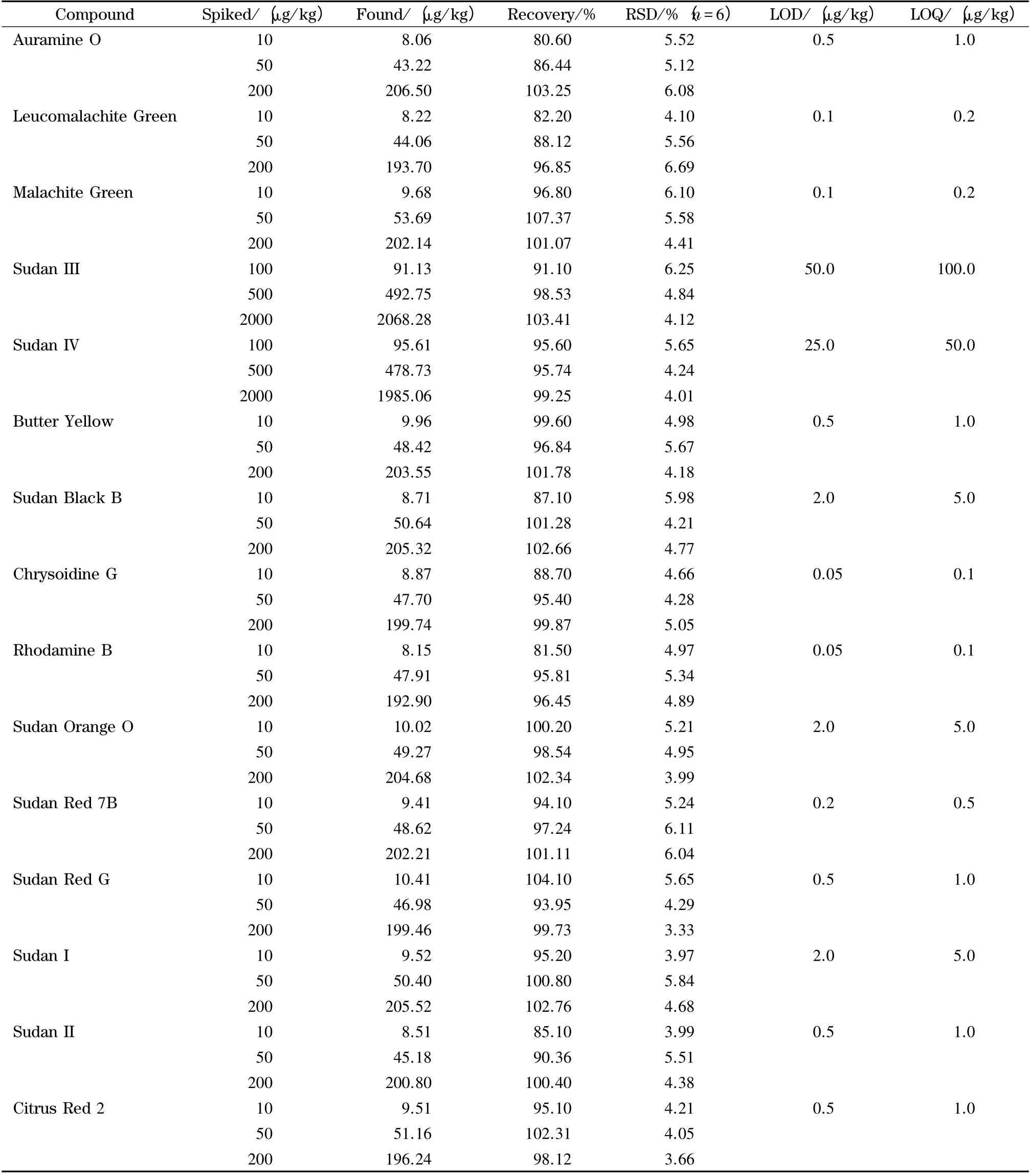
表4 15 种碱性合成色素的回收率、RSDs、检出限和定量限Table 4 Recoveries,RSDs,limits of detection (LODs)and limits of quantification (LOQs)of the 15 basic artificial dyes
2.3.3 方法应用
选取市售22 份水产品——白蟹、富贵虾、小黄鱼、大黄鱼、龙头鱼、黄姑鱼、梅童鱼、鱼免鱼、玉秃、鲳鱼、带鱼、青占鱼、马鲛鱼、鳓鱼、鲭鱼、鱿鱼、乌贼、海带、海蜇、海地瓜、蛏子和文蛤作为方法验证样品基质,并做了添加回收质控样(鲳鱼样品,模拟水平200 μg/kg),结果未发现阳性样品,质控样回收率均大于80%。下一步将以水产制品为采集对象,扩大筛查范围和样品种类。
将添加回收质控样以UPLC-Q-TOF MS 技术检测分析,得到总离子流图,对目标物进行相对分子质量及标准物质谱图数据库检索,结果可知15 种碱性合成色素化合物的匹配度均较高,其匹配度在85%以上。结果表明,经过乙腈和乙酸混合液提取和C18复合硅胶基质萃取管净化,15 种碱性合成色素能够被UPLC-Q-TOF MS 有效筛查出来。
3 结论
本文利用C18复合硅胶基质萃取管提取净化,结合UPLC-Q-TOF MS 技术,建立了水产品中15 种碱性合成色素的快速检测方法。样品前处理优化了提取剂和萃取柱种类的选择,对水产品中不同性质的合成色素进行了有效提取分离;并利用了Q-TOF MS 的精确质量数定性功能对不同种类的色素化合物进行筛查。与目前已报道的方法相比,前处理简便、快捷、高效,数据采集和数据库建立采用全息离子扫描的方式进行,所有二级碎片的精确质量和丰度都能用来匹配,适用于水产品中15 种碱性合成色素的快速筛选。
[1] Liu S J,Fan M,Jiang N,et al. Chinese Journal of Health Laboratory Technology (刘思洁,范明,姜楠,等. 中国卫生检验杂志),2013,23(12):2593
[2] Qu Z K,Deng G H,Wang S W,et al. Food Science (屈忠凯,邓光辉,王士伟,等. 食品科学),2015,36(4):217
[3] Zhang Z L,Mei Y J,Zhang Q M,et al. Chinese Journal of Analysis Laboratory(张贞理,梅英杰,张全美,等. 分析试验室),2012,31(4):15
[4] Tian X H,Yu Z Q,Chen W,et al. Food Science (田秀慧,于召强,陈玮,等. 食品科学),2013,34(12):171
[5] Zhu C Y,Wei J,Dong X F,et al. Chinese Journal of Chromatography (朱程云,魏杰,董雪芳,等. 色谱),2014,32(4):419
[6] Halme K,Lindfors E,Peltonen K. J Chromatogr B,2007,845:74
[7] Tao Y F,Chen D M,Chao X Q,et al. Food Control,2011,22:1246
[8] Jing S J,Li J Q. Journal of Shanxi Datong University:Natural Science (景顺杰,李建晴. 山西大同大学学报:自然科学版),2007,23(3):22
[9] Ding C H,Qian L,Ren H H,et al. Food Science (丁长河,钱林,任惠华,等. 食品科学),2007,28(2):244
[10] Deng G H,Chen S Y,Zhang G H. China Condiment (邓光辉,陈盛余,张桂华,等. 中国调味品),2011,36(4):92
[11] Song X,Ji S L,Yang L,et al. Chinese Journal of Food Hygiene (宋新,纪双利,杨丽,等. 中国食品卫生杂志),2009,21(5):422
[12] Xing W W,Wang R M,Wang J Q,et al. Chemical Research and Application (邢玮玮,王榕妹,王俊卿,等. 化学研究与应用),2010,22(1):42
[13] Zhang H,Chen D Y,Feng J L,et al. Chinese Journal of Health Laboratory Technology (张昊,陈东洋,冯家力,等.中国卫生检验杂志),2014,24(15):2167
[14] Yang L,Chen Q J,Ding X R,et al. Science and Technology of Food Industry (杨琳,陈青俊,丁献荣,等. 食品工业科技),2011,32(2):350
[15] Lin H L,Zhong B P,Hua Y Y. Chinese Journal of Health Laboratory Technology (林宏琳,钟碧萍,华永有. 中国卫生检验杂志),2014,24(2):156
[16] Liu M,Li X L,Bie W,et al. Chinese Journal of Chromatography (刘敏,李小林,别玮,等. 色谱),2011,29(2):162
[17] Chen Y,Hua J,Pan Y L,et al. China Condiment(陈勇,花锦,潘亚利,等. 中国调味品),2014,39(4):124
[18] Lin D Q,Wan C B,Qiu P,et al. Journal of Chinese Mass Spectrometry Society (林黛琴,万承波,邱萍,等. 质谱学报),2013,34(3):171
[19] Zhao Y S,Yang M L,Zhang F,et al. Chinese Journal of Chromatography (赵延胜,杨敏莉,张峰,等. 色谱),2011,29(7):631
[20] Lü D M,Ding Y L,Zhan S,et al. Modern Instruments &Medical Treatment(吕东明,丁云连,詹晟,等. 现代仪器与医疗),2013,19(2):52
猜你喜欢
食品安全导刊(2020年14期)2020-12-04 20:19:39
现代食品(2018年16期)2018-11-02 02:33:52
健康博览(2017年12期)2018-02-06 21:30:06
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:45
中成药(2017年4期)2017-05-17 06:09:46
食品工业科技(2016年17期)2016-10-31 02:45:08
浙江农业科学(2016年11期)2016-05-04 04:16:45
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
核科学与工程(2015年3期)2015-09-26 11:58:24
电源技术(2015年2期)2015-08-22 11:27:56