QuEChERS-液相色谱-质谱法快速筛查和确证大米中205 种农药残留
2015-12-26 01:59:20曲世超黄大亮刘佳成贾彦波纪明山
色谱 2015年10期
陈 溪, 程 磊, 曲世超, 黄大亮, 刘佳成,崔 晗, 贾彦波, 纪明山
(1. 沈阳农业大学植物保护学院,辽宁 沈阳110866;2. 大连出入境检验检疫局检验检疫技术中心,辽宁 庄河116400;3. 上海爱博才思分析仪器贸易有限公司,北京100015)
我国是世界上最大的水稻生产国之一,全国近60%的人口以大米为主食。我国也是大米出口国,自1994 年以来,每年出口大米200 多万吨,主要出口日本、韩国和非洲一些国家。农药的使用可以提高农产品的产量和品质,但随着其广泛使用,农药残留问题已引起社会的广泛关注,国际社会设置了严格的限量标准。国际食品法典委员会(CAC)规定粮谷中的最高农药残留限量(MRL)为0.02 ~20.0 mg/kg;我国于2014 年实施了国家标准GB 2763-2014《食品中农药最大残留限量》,涉及农药387种、检测项目3 650 项[1]。目前,出口至韩国的大米农残检验项目已扩增到293 项,出口至日本的大米农残检验项目扩增到581 项。我国对于大米样品的检测常采用GB/T 20770-2008,采用该方法通常不能得到用来确证的质量较好的二级质谱图,发现疑似阳性样品时需要重新设定方法,将样品重新分析测定一次,影响效率并增加了工作量。因此建立大米中农药多残留的快速筛查和确证方法具有重要意义。
目前,用于农药多残留检测的前处理技术已有多篇综述类文献报道,主要有超临界流体萃取(SFE)、加速溶剂萃取(ASE)、微波辅助萃取(MAE)、固相萃取(SPE)、固相微萃取(SPME)、凝胶渗透色谱(GPC)、基质固相分散萃取(MSPD)、分子印迹(MIP)和QuEChERS 等技术[2-6],其中QuEChERS 因简便、快速、安全、高效等优点而应用广泛。农药多残留的检测方法主要有酶联免疫分析法(ELISA)[7-9]、气相色谱法(GC)[10]、气相色谱-质谱法(GC-MS)[11-13]、气相色谱-串联质谱法(GCMS/MS)[14-17]、液相色谱法(LC)[18,19]、液相色谱-质谱法(LC-MS)[20,21]、液相色谱-串联质谱法(LCMS/MS)[22-28]等,其中LC-MS/MS 的灵敏度和准确性高,应用广泛。
液相色谱-三重四极杆复合线性离子阱质谱(LC-Q-TRAP/MS)具有多反应监测-信息关联采集-增强子离子扫描(MRM-IDA-EPI)检测模式,能够对未知化合物进行双重定性,大大排除了假阳性结果,提高了定性分析的准确度;并且能够利用高选择性和高灵敏度的s-MRM 扫描获得化合物的峰面积信息,对化合物进行定量分析。本文结合QuEChERS和LC-MS/MS 建立了大米中205 种农药的快速筛查分析方法,与传统的目标化农药残留检测方法相比,该方法适合对食品中的非目标农药进行扫描和鉴定,并通过二级谱库检索进一步确证。该方法能够一次进样同时检测大米中杀虫剂、杀螨剂、除草剂、杀线虫剂、杀菌剂、生长调节剂等各种农药,比GB/T 20770-2008 扩增了74 个检测项目,并且具有快速、准确、灵敏度高等特点,不用分组便能准确进行定性和定量分析,对大米样品进行实际检测仅需20 min。
1 实验部分
1.1 仪器与试剂
AB SCIEX 5500 QTRAP 三重四极杆复合线性离子阱质谱系统(美国Applied Biosystems 公司);Shimadzu HPLCXR系统(日本岛津公司);冷冻离心机3K15(德国Sigma 公司);旋涡混合器MS3(德国IKA 公司)。
乙腈、甲醇(HPLC 级,德国Merck 公司);甲酸、甲酸铵、乙酸(HPLC 级,上海安谱科学仪器有限公司);PSA、C18吸附剂(美国Agilent 公司);聚丙烯离心管15 mL、50 mL(德国Greiner 公司);滤膜0.2 μm Econofilters PTFE(聚四氟乙烯)(美国Agilent 公司);GSB(石墨化炭黑,美国Supelco 公司);无水MgSO4、无水CH3COONa、无水Na2SO4(分析纯,科密欧公司);高纯水(经Milli-Q 超纯水器纯化得到)。标准品购自兰伯瑞公司(A ~H,J,K 10 组,共计205 种农药),质量浓度为100 mg/L,纯度均大于98.3%。
1.2 标准溶液的配制
从上述10 组质量浓度为100 mg/L 的混合标准乙腈溶液中各取10 μL,用900 μL 乙腈稀释,制备质量浓度为1 mg/L 的储备液,于-18 ℃冰箱中保存。用空白大米样品基质液作为溶剂,将上述储备液逐级稀释制备标准工作曲线溶液,质量浓度分别为0.1、0.25、0.5、2、5、10、15、20 μg/L。
1.3 样品前处理
按照GB 5491-1985 扦取的大米样品经过粉碎机粉碎,过20 目筛,混匀,密封作为试样。精确称取5.00 g 样品于50 mL 离心管中,加入10 mL 水,涡旋混合1 min;加入乙腈(含0.1% (v/v)乙酸)15 mL,涡旋1 min;加入6 g 无水MgSO4与1.5 g CH3COONa,剧烈振摇20 s 至块状结晶散开,4 000 r/min 离心5 min。取上清液8 mL 于预先加有400 mg PSA、1 200 mg 无水MgSO4及400 mg C18吸附剂的15 mL 离心管中,涡旋混合15 min,4 000 r/min 离心5 min,上清液过0.2 μm 的滤膜后,供LC-Q-TRAP/MS 测定。
1.4 色谱条件
色谱柱:Phenomenex Synergi Fusion-RP C18(50 mm×2.0 mm,粒径2.5 μm,孔径10 nm);柱温:40 ℃;流动相:A 相为5 mmol/L 甲酸铵水溶液(将5 mL 1 mol/L 甲酸铵溶液与895 mL 水、100 mL 甲醇混合超声即得);B 相为5 mmol/L 甲酸铵甲醇溶液(将5 mL 1 mol/L 甲酸铵溶液与95 mL水、900 mL 甲醇混合超声即得);流速:0.4 mL/min;梯度洗脱程序:0 ~1.0 min,100%A;1.0~15.0 min,100% A ~100% B;15.0 ~18.0 min,100%B;18.0~18.1 min,100%B~100%A;20 min完成梯度洗脱程序。进样量:10 μL。
1.5 质谱条件
离子源温度:550 ℃;扫描模式:正离子模式;离子化电压(IS):+5 500 V;气帘气(CUR)压力:241.3 kPa;MRM-IDA-EPI 检测模式;喷雾气(GS1)压力:379.2 kPa;辅助加热气(GS2)压力:413.7 kPa;接口的加热器:On;碰撞气(CAD)压力:High;智能MRMTM时间监测窗口:120 s;目标扫描时间:0.7 s;入口电压(EP):10 V;出口电压(CXP):13 V;其他参数见表1。
2 结果与讨论
2.1 QuEChERS 前处理方法的优化
QuEChERS 是一种快速、简单、廉价、高效、耐用、安全的样品前处理方法,参考AOAC 2007.01[29]和EN 15662[30]两种方法对盐析剂和吸水剂进行选择。常用的盐析剂为NaCl 和CH3COONa,吸水剂为无水Na2SO4和无水MgSO4,本文分别对此进行了比较分析。对于两种盐析剂,结果表明NaCl 和CH3COONa 对回收率没有显著的影响,平均回收率相差在3.0%以内。苯噻草胺是除草剂中的噻唑多环化合物,双硫磷是有机磷类杀虫剂,咪唑菌酮为含苯胺基的杀菌剂,绿麦隆为含苯甲基除草剂,西草净为三嗪类芽前除草剂,嘧菌酯为甲氧基丙烯酸酯类杀菌剂,这些化合物能代表大多数化合物的结构特点,因此在10 μg/L 添加水平下,本文以上述6 种农药为考察对象,考察了无水MgSO4和无水硫酸钠两种吸水剂对农药提取效果的影响。结果表明,使用无水MgSO4的回收率要优于无水硫酸钠,回收率提高了0.9%~31.7%。原因在于净化步骤中使用的PSA 属于正相吸附剂,样品含水量越少净化效果越好,无水MgSO4的吸水能力强且在吸水过程中放热,有利于农药的提取;而且无水MgSO4粒度较小,在振摇和涡旋过程中与样品的混合更加充分,并能够与乙腈发生协同作用从而提高提取效率。另外,QuEChERS 方法在净化过程中,常采用PSA 吸附剂除去样品基质中的有机酸、色素、糖、脂肪酸等,GCB去除甾体、叶绿素、类胡萝卜素等,C18吸附剂除去基质中的脂肪和脂类等非极性有机物,因此在净化过程中仅添加了PSA 和C18吸附剂用于吸附大米样品中的脂肪酸、氨基酸、脂肪和脂类等有机物。
2.2 色谱条件的优化
2.2.1 流动相的选择
电喷雾质谱中样品的电离是在溶液状态下进行的,因此流动相的组成和添加剂不仅影响分析物的色谱保留时间和峰形,还会影响分析物的离子化效率,从而影响目标化合物的检测灵敏度。由于质谱检测要求流动相的挥发性能好,而且甲醇是质子性溶剂,有利于样品离子化,因此本文考察了5 mmol/L 甲酸铵水溶液与5 mmol/L 甲酸铵甲醇溶液二元体系、甲醇-1%(v/v)甲酸水溶液和甲醇水溶液分别作为流动相时对农药残留的分离效果。对10 μg/L 的205 种农药混合标准溶液进行分析,发现仅以甲醇水或者添加1%(v/v)甲酸作为流动相时,样品中许多组分有峰拖尾现象,且霜霉威、灭蝇胺、呋虫胺等农药尤为严重,原因可能是流动相的pH 值不适合农药组分的分离。而采用5 mmol/L甲酸铵水溶液与5 mmol/L 甲酸铵甲醇溶液作为流动相时,205 种农药的响应较好;进一步调节流动相的比例,205 种农药都能得到较好的分离。因此选用5 mmol/L 甲酸铵水溶液与5 mmol/L 甲酸铵甲醇溶液作为流动相。
2.2.2 色谱柱的选择
根据待测农药性质,一般选择C18色谱柱。本文共考察了4 种色谱柱:1 号色谱柱Phenomenex Synergi Fusion-RP C18柱(50 mm×2.0 mm,2.5 μm);2 号色谱柱Waters ACQUITY UPLC BEN C18柱(50 mm×2.1 mm,1.7 μm);3 号色谱柱Phenomenex Synergi Fusion-RP C18柱(50 mm×2.0 mm,4 μm);4 号色谱柱RESTEK Ultra AQ C18柱(100 mm×2.1 mm,3 μm)。实验发现采用1号色谱柱可以实现205 种农药较好的分离,且峰形较好;2 号色谱柱分析时间长,且峰形较差;3 号色谱柱分离不显著;4 号色谱柱分析时间较长。考虑到快速分析的要求,本文最终选择1 号色谱柱。
2.3 质谱条件的优化
质谱检测的各种条件也是影响分析结果的重要因素之一。本文首先通过全扫描确定各农药的分子离子与特征碎片离子,然后对离子源温度、喷雾器、辅助加热气等操作参数进行了系统的优化,结果表明,当离子源温度为550 ℃、喷雾器压力为379.2 kPa、辅助加热气压力为413.7 kPa、气帘气压力为241.3 kPa、离子化电压为+5 500 V 时,样液中所有农药组分的离子化效率和质谱响应信号均达到最佳。最终,在上述最佳的流动相、色谱柱以及质谱条件下确定了各目标物的定量离子、辅助定性离子、去簇电压(DP)和碰撞电压(CE)(见表1)。205 种农药混合标准溶液在MRM 模式下的总离子流图见图1。
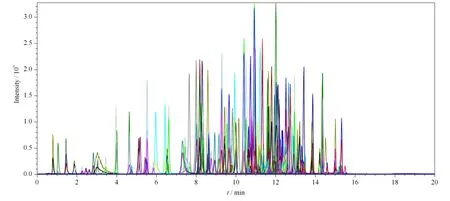
图1 质量浓度均为5 μg/L 的205 种农药混合标准溶液在MRM 模式下的总离子流图Fig.1 Total ion chromatogram for a mixed standard solution of the 205 pesticides (5 μg/L for each)in MRM mode

表1 205 种农药的保留时间、定量及定性离子对、DP、CE、平均回收率和相对标准偏差(RSD)(n=6)Table 1 Retention times,quantitative and qualitative ion pairs,declustering potentials (DP),collision energies (CE),average recoveries and relative standard deviations (RSDs)of the 205 pesticides (n=6)
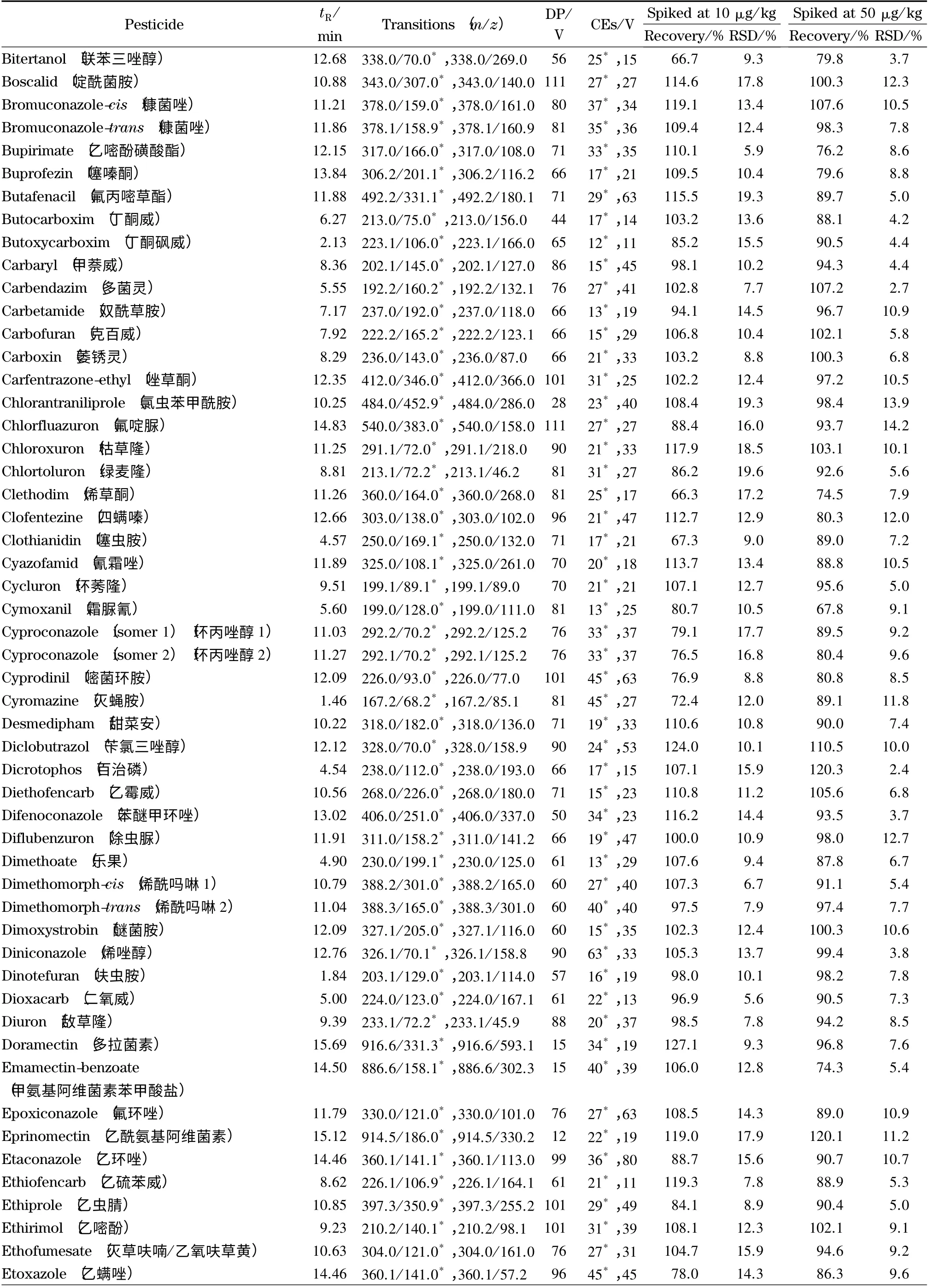
表1 (续)Table 1 (Continued)

表1 (续)Table 1 (Continued)
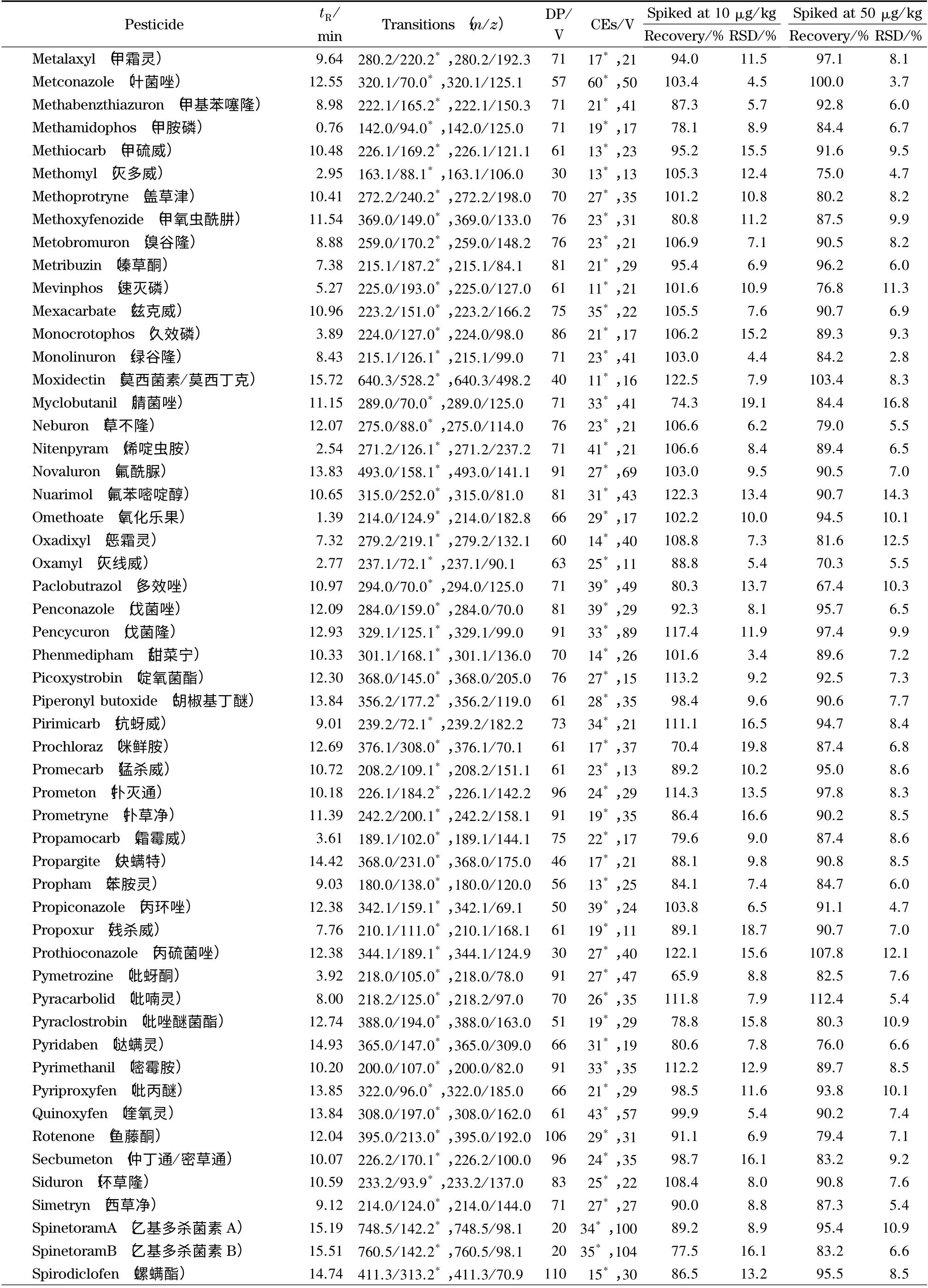
表1 (续)Table 1 (Continued)
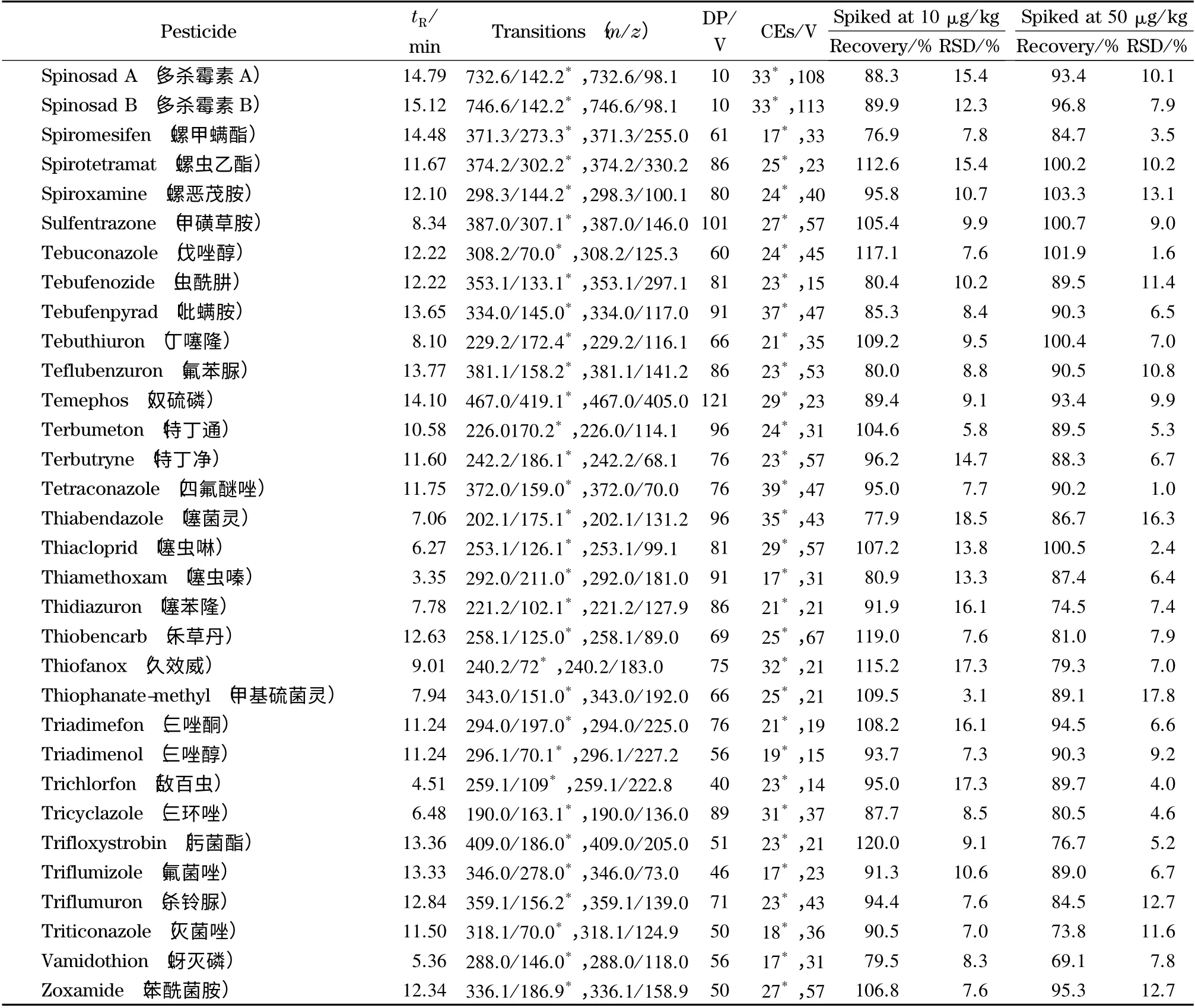
表1 (续)Table 1 (Continued)
2.4 质谱扫描方式的选择及谱库的检索
常规的串联四极杆质谱只能得到MRM 的两对离子的色谱峰,定性仅靠两对离子的丰度比确定;本研究使用的仪器是LC-Q-TRAP/MS,并采用了MRM-IDA-EPI 扫描模式,由于该方法使用的是高灵敏度的增强型二级谱库,谱库中最少含有4 张谱图,包括高、中、低3 个能量以及(35±15)V 的平均图,因此对标准溶液的标准谱图匹配度较高(均在85%以上)。另外,该谱库是开放性的,能够在原有谱库的基础上,将205 种农药的EPI 图谱补全,进而实现一次进样同时得到MRM 的色谱图和相应高灵敏度的EPI 图,并能够通过保留时间、碎片离子对及高灵敏度的EPI 图进行定性、定量分析,有效地降低了假阳性结果。
2.5 方法学验证
2.5.1 方法的线性范围、相关系数及定量限
用空白大米样品基质液作为溶剂制备标准工作曲线溶液,按1.4 节及1.5 节的条件进行测定,以目标物的质量浓度x (μg/L)为横坐标,定量离子的峰面积y 为纵坐标绘制工作曲线。结果显示,3-羟基克百威等178 种农药的线性范围为0.1 ~20.0 μg/L;阿维菌素、多拉菌素、甲氨基阿维菌素苯甲酸盐、乙酰氨基阿维菌素、氟虫脲、氟铃脲、伊维菌素、氰氟虫腙、莫西菌素、氟酰脲、乙基多杀菌素和多杀霉素12 种农药的线性范围为2.0~20.0 μg/L;氟啶脲、氟啶胺和双硫磷3 种农药的线性范围为1.0 ~20.0 μg/L;环丙唑醇及水胺硫磷的线性范围为0.25~20.0 μg/L;乙环唑、乙螨唑、喹螨醚、唑螨酯、氟虫腈、氟蚁腙、茚虫威、虱螨脲、哒螨灵和螺甲螨酯10 种农药的线性范围为0.5 ~20.0 μg/L。所有农药的线性相关系数均大于0.995。以10 倍信噪比(S/N)确定农药的定量限,205 种农药的定量限均低于10μg/kg,满足农药残留限量的要求,其中176种农药的定量限为0.5 μg/kg,3 种农药的定量限为5 μg/kg,11 种农药的定量限为2.5 μg/kg,2 种农药的定量限为1.25 μg/kg,13 种农药的定量限为10.0 μg/kg。
2.5.2 方法的回收率和精密度
采用QuEChERS 方法对大米样品进行提取净化,添加水平为10 μg/kg 时,205 种农药的平均回收率范围为62.4%~127.1%,相对标准偏差(RSD)范围为1.9%~20.0%,其中RSD≤10%的有90 种,10%<RSD<20%的有115 种;添加水平为50 μg/kg时,平均回收率范围为65.9%~120.3%,RSD 范围为1.0%~18.4%,其中RSD≤10%的有125 种,10%<RSD<20%的有80 种(见表1)。该研究方法可以达到检测低含量多农药残留的要求。
2.6 实际大米样品中205 种农药残留的分析检测
将建立的方法用于市场上购买的3 种东北大米、1 种盘锦大米、1 种泰国大米和1 种五常大米中205 种农药残留的快速筛查分析,在MRM-IDA-EPI扫描模式下通过对205 种农药的筛查分析,发现有两种农药疑似阳性,对其进行谱库检索进一步判定未知农药,通过查看未知化合物增强型二级全扫描谱,并与标准样品的二级全扫描谱库匹配,结果显示在6 个实际样品中,有一个样品检出了三环唑,与标准谱库匹配率为94.956%,测定值为5.70 μg/kg;同时该样品还检出多菌灵,与标准谱库匹配率为89.261%,测定值为2.78 μg/kg。在中国的水稻生产过程中,三环唑与多菌灵是应用较广的杀菌剂,对防止稻瘟病与胡麻叶斑病具有较好的效果,每年多次使用,若在喷洒农药过程中没有注意安全间隔期,可能会导致残留。将检出的大米样品与中国、日本、韩国、欧盟的最大残留限量(MRLs)要求进行了比对,可以看出该检测值远远低于各国最大残留限量要求(见表2)。该检测结果进一步说明了本文所建立的大米中农药残留快速筛查和确证方法能够快速、高灵敏的对农药残留进行分析检测。
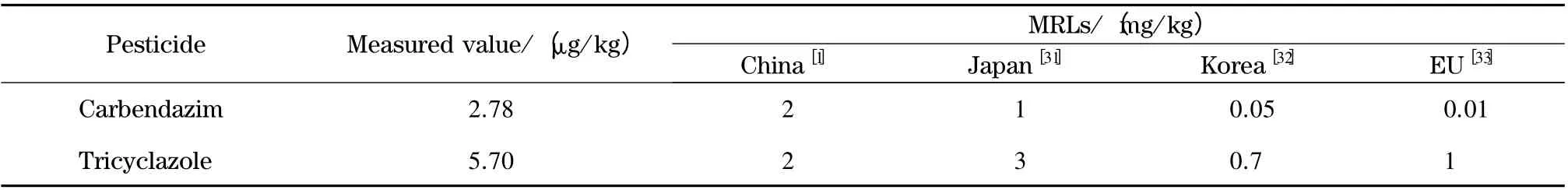
表2 大米样品中检出的农药含量和各国最大农药残留限量(MRLs)Table 2 Pesticides measured in one rice sample and their MRLs
3 结论
本文采用LC-MS/MS 技术建立了大米中205种农药残留的快速筛查分析方法,涵盖了出口日本的大米农药残留检验的136 个检测项目和出口韩国的大米农药残留检验的88 个检测项目。与GB/T 20770-2008 相比具有以下区别及优势:一是本方法涵盖了标准中的131 个检测项目,扩增了74 个检测项目。二是标准中采用的是传统的三重四极杆串联质谱法,将486 种农药分为7 组,每组的分析时间为23 min;而本方法仅需一次进样即可检测205 种农药,且分析时间仅需20 min。三是标准中采用两对MRM 及比值定性,判断是否含有该农药;而本方法采用MRM-IDA-EPI 扫描模式,一次进样即可同时得到MRM 和母离子所有高灵敏度的二级碎片,通过谱库检索排除假阳性结果。综上所述,本文建立的方法快速、准确、灵敏度高、筛查范围广,适用于大米中农药残留的快速、全面筛查分析,为大米进行风险监测提供了一种有力的技术手段,可为食品安全监管提供有效的保障。
[1] GB 2763-2014
[2] Dimitra A L,Triantafyllos A A. Anal Bioanal Chem,2007,389:1663
[3] Angelika B,Marek B. Food Chem,2008,108(2):669
[4] Yolanda F,Alexander J K,Jon W W. J Agric Food Chem,2010,58(10):5859
[5] Chen J,Duan C F,Guan Y F. J Chromatogr B,2010,878(17/18):1216
[6] Monika K,Marek B. Trends Anal Chem,2010,29(9):1064
[7] Zhang R,Liu K C,Cui Y L,et al. RSC Adv,2015,5:35874
[8] Chen Y N,Kong D Z,Liu L Q,et al. Anal Methods,2015,7:3559
[9] Yan X,Li H X,Yan Y,et al. Anal Methods,2014,6:3543
[10] Jin B H,Xie L Q,Wu W D,et al. Chinese Journal of Analysis Laboratory (靳保辉,谢丽琪,吴卫东,等. 分析试验室),2008,27(6):75
[11] Zhang W G,Chu X G,Li C J,et al. Chinese Journal of Analytical Chemistry (张伟国,储晓刚,李重九,等. 分析化学),2006,34(4):484
[12] Nguyen T D,Han E M,Seo M S,et al. Anal Chim Acta,2008,619(1):67
[13] GB/T 19649-2006
[14] Hernández F,Cervera M I,Portolés T,et al. Anal Methods,2013,5:5875
[15] Hou X,Han M,Dai X H,et al. Food Chem,2012,138(2/3):1198
[16] Cheng Z,Zhang R,Liu W H,et al. Chinese Journal of Chromatography (程志,张蓉,刘韦华,等. 色谱),2014,32(1):57
[17] Ma Z L,Zhao W,Li L Y,et al. Chinese Journal of Chromatography (马智玲,赵文,李凌云,等. 色谱),2013,31(3):228
[18] Qi R F,Jiang H C,Liu S X,et al. Anal Methods,2014,6:1427
[19] Jitlada V,Rodjana B. Anal Methods,2012,4:2101
[20] Sami B,Olivier P,Marie-Florence G L. Anal Bioanal Chem,2003,376:355
[21] Sami B,Olivier P,Marie-Florence G L. Anal Bioanal Chem,2003,376:157
[22] GB/T 20770-2008
[23] Sung J L,Hyeong J P,Wooseong K,et al. Biomed Chromatogr,2009,23(4):434
[24] Zahra D,Mohammad K R,Syed W H. Anal Methods,2013,5:1192
[25] Kang J,Fan C L,Chang Q Y,et al. Anal Methods,2014,6:6285
[26] Li J C,Liu H,Yu M Q,et al. Anal Methods,2014,6:9124
[27] Gao J Y,Wang J,Zuo M,et al. RSC Adv,2015,5:5895
[28] Zheng S N,Li L Y,Lin H,et al. Chinese Journal of Chromatography (郑姝宁,李凌云,林桓,等. 色谱),2013,31(1):71
[29] AOAC Official Method 2007.01
[30] BS EN 15662-2008
[31] The Japan Food Chemical Research Foundation. Maximum Residue Limits(MRLs)of Agricultural Chemicals in Foods.(2015-03-26)[2015-04-15]. http://www.m5.ws001.squarestart.ne.jp/foundation/fooddtl.php?f_inq=100
[32] Korea Food &Drug Administration. MRLs for Pesticides in Foods-2012. 2. (2012-02-24). http://down.foodmate.net/standard/sort/13/28919.html
[33] Commission Regulation (EC)No 149/2008
猜你喜欢
今日农业(2021年17期)2021-11-26 23:38:44
今日农业(2021年14期)2021-11-25 23:57:29
食品安全导刊(2021年20期)2021-08-30 06:39:48
今日农业(2021年9期)2021-07-28 07:08:42
今日农业(2019年16期)2019-01-03 11:39:20
中国蜂业(2018年4期)2018-05-09 06:25:08
当代化工研究(2016年6期)2016-03-20 16:21:46
当代化工研究(2016年5期)2016-03-20 16:21:35
特产研究(2014年4期)2014-04-10 12:54:22
Sciences in Cold and Arid Regions(2014年6期)2014-03-31 00:28:31