
生物信息学在微生物生态学中的应用
2015-12-26 01:08李靖宇
安徽农业科学 2015年22期
生物信息学在微生物生态学中的应用
李靖宇
(北方民族大学生物科学与工程学院,宁夏银川 750021)
摘要高通量测序技术的快速发展极大地推动了微生物多样性的研究,各种生物信息学软件的开发用于分析由此产生的大量基因信息。综述了生物信息学在微生物生态学中的应用以及相关软件和数据库在α、β-多样性研究中的应用。
关键词生物信息学;微生物生态学;应用
中图分类号S188;X172
基金项目北方民族大学引进人才科研启动项目(44/4400302502)。
作者简介李靖宇(1986-),男,内蒙古包头人,讲师,从事环境微生物学研究。
收稿日期2015-06-11
Application of Bioinformatics in Microbial Ecology
LI Jing-yu (College of Biological Science & Engineering, Beifang University of Nationality, Yinchuan, Ningxia 750021)
AbstractThe rapid development of high-throughput sequencing technology has greatly promoted the study of microbial diversity, and various development of bioinformatics software was used to analyze a large number of genes generated by using this technology. This paper summarizes application of bioinformatics in microbial ecology and their corresponding software and synthesis databases used in α, β-diversity studies.
Key words Bioinformatics; Microbial ecology; Application
微生物生态学的主要任务之一是研究不同环境中微生物类群的组成、结构以及生态功能。其中多样性研究是一项重要的内容,目前的研究手段主要是基于可培养的方法和基于DNA技术的分子生物学方法。后者粗略地可分为指纹图谱技术与测序技术两大类。所有这些技术或方法的建立,都是为了解决微生物生态学的一个核心问题,最大限度地描述自然环境中微生物的群落组成或者是群落结构的时空动态变化过程。在描述的环节中,需要获得一些具体参数,直观地表现自然环境中微生物群落的基本情况。一般从以下几个方面来描述,主要有特定环境中微生物各类群的组成情况、相对丰度,多样性指数(如Shannon、Simpson指数等)以及估计物种总数的指数(如Richness、ACE、Chao1),物种亲缘关系分析,分类学鉴定等内容。基于不同技术对微生物多样性,即通过指纹图谱技术或测序技术来描述,后续所需要的分析手段不同。这2种技术获得数据主要以图谱的形式和基因序列的形式呈现,特别是近几年高通量测序技术的快速发展,产生了大量的基因序列信息,虽然可以人工处理这些数据,但费时费力、易产生人为错误等,这就需要引入新的、高效的分析手段,因此,生物信息学在微生物生态学研究中的应用受到了广泛关注。
1 生物信息学软件概述
针对微生物生态研究的技术手段,后续分析的各种生物信息学软件快速发展,主要包括基础分析的软件,如ClusterW、CLC Sequence Viewer、BioEdit、ARB等;亲缘关系分析的软件,如Mage、Phylip、PAUP、ARB等;指纹图谱分析软件,如Quantity One、T-Alin、T-REX等;群落结构比较的软件,如Libshuff、∫- Libshuff 、UniFrac、Fast-UniFrac等;多样性指数分析软件,如DOTUR等;序列提交的软件,如Sequin、BankIt、Sequence Read Archive等;高通量数据/综合分析软件,如Mothur、Qiime、RDP Pipeline等。基于培养或基于DNA分子技术获得微生物物种基因信息,可以在NCBI/EMBL/DDBJ、RDP以及Silva数据库中进行比对、分类鉴定(图1)[1-21]。
这些软件是基于Pyton、C++、Java、PHP、Perl等语言编写的,可以在线使用,也可以下载使用,具体要求针对不同软件而有所不同,大多数软件可以在Windows、Linux、Mac OS 等系统下安装与运行,有一些软件在Windows下无法安装与运行,需要在Windows系统上安装Virtual Box来运行这些软件,如Qiime软件。但这些软件都带有详细、具体的使用操作说明,所以很容易学习与使用[1-21]。
2生物信息学软件的应用
生物信息学软件的进步提高了解释相关微生物生态学问题的能力。伴随着软件,如ARB、DOTUR、LIBSHUFF、UniFrac、HOMOVA、AMOVA、TreeClimber以及rRNA数据库的快速发展(表1),微生物生态学已经从描述性阶段发展到试验尝试阶段。虽然这些工具已广泛使用,但随着测序能力的不断提高以及研究内容越来越复杂,这些软件还有许多局限之处。首先,对于一些容易使用的rRNA数据库拥有在线分析工具,主要包括序列比对、基因信息物种分类、分析流程;然而,这些工具只能做有限的一般性分析,值得疑问的是把巨大的数据集通过互联网进行分析是不是一个可持续的实践。第二,许多现有软件的开发仅适用于分析102~104条序列。随着序列数量的不断增多,至关重要的是把现有的软件进行重构,使用更高效的算法。此外,尽管已经使用脚本语言,如Perl和Python用于在线分析较小的数据集,但它们相比用C和C++编写的代码在运行速度上慢得多。最后,现有软件的特征限制了他们的集成和进一步发展。随着测序能力的不断提高以及研究内容越来越复杂,关键的问题是软件的开发要灵活和易于维护。Mothur是美国密歇根大学的Patrick D. Schloss在2009年开发的一个独立的数据处理平台,它的前身是Dothur软件。Mothur软件使用现代的面向对象的编程策略,用C++编写,无需安装,在Windows、Linux和Mac OS系统上都可以运行。它整合了Pyrosequencing pipeline (RDP)、NAST、SINA、RDP aligners、DNADIST、DOTUR 、CD-HIT、SONS、TreeClimber、∫- Libshuff 和UniFrac,在运算法则上做了一些改进,使得Mothur这个软件更加灵活、速度更快、资源利用效率更高。但Mothur不能够直接出图,必须依赖于其他软件,如R语言。除罗氏454数据处理外,Mothur目前也有了针对Illumina数据的处理方式,从Mothur官方网页上即可以看到Patrick D. Schloss写的标准数据处理流程[1]。
另一个被广泛使用的高通量数据处理平台是QIIME(Quantitative Insights Into Microbial Ecology),是美国科罗拉多Rob Knight及其开发团队等于2009年开发出来的。QIIME只能在Mac OS和Linux系统上运行,也可以通过在Windows系统上安装Virtual Box来运行。QIIME支持多种微生物群落结构的分析与可视化,包括网络分析、样品内或样品间多样性的直方图以及分析是否在特定的栖息地存在具有代表性的“核心”微生物类群。QIIME还提供了允许用户与数据进行交互的图形化显示。软件的实现是高度模块化的,并大量使用单元测试确保结果的准确性。这种调整可以使具有其他功能的选择性组件,如选择操作分类单元,序列比对,推断系统发育树和基于系统发育和分类单元的样品内及样品间多样性分析(包括第三方应用程序的整合)变得容易被集成与整合[2]。
无论使用哪个软件,都可以很好地处理细菌16S rRNA的高通量测序数据,因为Mothur和QIIME都包含了细菌16S rRNA比对和分类的数据库。如果是古菌16SrRNA的数据,虽然RDP、Greengenes、SILVA等数据库可以用来分析多样性,但分类效果较差。因为古菌的纯培养很少,分类仍处于发展阶段。如果是功能基因,就面临更大的难题。数据处理的难点主要有:第一,比对没有可供使用的参考数据库;第二,分类需要序列经过比对之后,分成不同的OTU,然后从每个OTU中选择一个代表序列,通过BLAST进行分类。鉴于这些缺陷与不足,目前对于研究功能基因的微生物生态学问题,可以使用基因芯片(microarray)以及DGGE或者是T-RFLP等方法[1-2]。
末端限制性片段长度多态性(T-RFLP)是研究微生物群落的另一种高通量技术,可以产生大量复杂的数据集。尽管在不断地完善与改进,这些数据的分析还是需要很多步骤和数据操作才能解释这些结果。这些步骤通常成为数据分析的障碍,浪费时间,且容易引入人为的分析错误。目前,分析数据的最大障碍主要有:第一,真伪峰的区分;第二,所有样品中峰值的比对;第三,从原始数据创建一个关于样品的二维数据矩阵;第四,数据矩阵的快速处理;第五,确定哪一种多变量分析更适合某一特定的数据集。为了克服这些障碍,可以通过T-REX、TAP-TRFLP、MiCA、PAT、TRAMPR对T-RFLP技术产生的数据进行有效的分析[3-7]。但这些程序之间的特性和功能差别很大,大多数程序最初并不主要是用来分析T-RFLP数据的。T-REX是一个免费的、基于Web的工具,只需通过Web浏览器联网即可对T-RFLP数据进行分析[7]。这个程序的具体使用在官网上有详细的操作流程,其中对数据处理执行特定功能的菜单按钮在很大程度上是独立的,可以在任何时候使用任何一个按钮,而不需要重新加载或上传相同的数据。
芯片技术需要对大量的数据进行有效地组织和分析。这些数据包括样品杂交的信息、杂交图像及从中提取的数据矩阵以及物理阵列、特征和报告分子的相关信息。有许多独立的软件系统,可以解决芯片数据处理与分析的一些需求,如用于芯片数据聚类与可视化的数据库和应用程序、包含报告分子信息的公共数据库、商业化的实验室信息管理系统(LIMS)以及各种用于记录生物材料注释的存储方法。然而,没有一个统一的系统能够有效地组织微阵列试验产生的所有信息以及用工具把这些信息进行整合用于微阵列杂交数据的量化分析。为了满足这些需求,BASE(BioArray Software Environment)提供了一个用于存储和分析微阵列信息的集成框架[8-9]。BASE是一个MIAME(Minimum Information About a Microarray Experiment guidelines)支持的用户自定义数据库和分析平台,可以在任何芯片实验室中进行安装并通过网络为许多用户提供同步服务。该软件是在GNU / Linux的操作系统上用PHP语言开发的,数据被存储在一个相关的数据库(MySQL)中,可以通过Apache Web服务器传输给用户。如有需要,用户界面可以使用Java以及JavaScript,且C++已经被用于在服务器上计算密集型的任务。总之,这个系统集成了生物材料的信息、原始图像以及数据提取的功能,并提供了一个插件框架用于数据转换、数据查看以及分析模块[8-9]。PhyloChip是一种用于微生物调查的16S rRNA基因芯片,已成功用于研究一些有趣环境中的微生物多样性。然而,它的使用受分析软件缺乏的限制。 PhyloTrac解决了这一需求,是第一款提供PhyloChip全程分析的桌面应用程序。通过多种集成显示提供了丰富的、交互式可视化结果。 PhyloTrac是免费的,通过整合PhyloChip芯片平台与专用分析应用程序为微生物调查提供了一个全面和易于使用的工具包[9]。
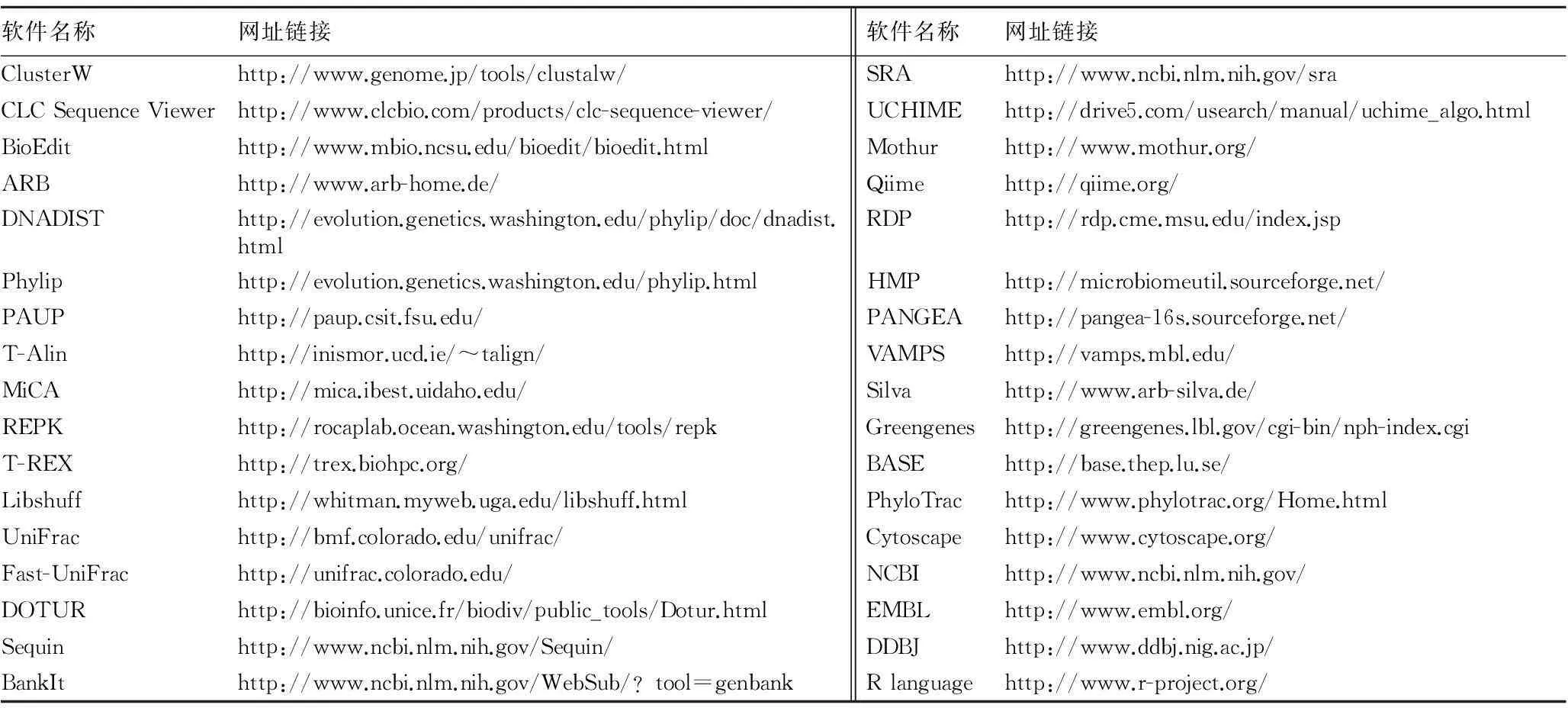
表1 常用软件汇总
3 基于α、β-多样性的微生物生态学研究
微生物生态学研究中,α、β-多样性研究尤为重要。α-多样性主要是反映一个独立群落中微生物种类的多少,度量方法主要包括Shannon、Simpson、Richness、ACE、Chao1等;β-多样性测量时间或空间上微生物群落之间的差异,即使在相同类型的环境中取多个样品得到的微生物群落也会有很大差异,目前应用最普遍的度量方法是相似性(相异性)指数度量群落物种组成在时空尺度上的变化。根据数据的特征,可供选择的指数很多,主要有用于二元数据的Sφrensen指数、Jaccard指数,用于数量数据的Bray-Curtis指数等。β-多样性计算可依赖于分类作为基础(如物种、属或者是OTUs的重叠部分)或者依赖于亲缘关系作为基础(如进化树中的重叠部分)。亲缘关系β-多样性测量与基于分类计算的β-多样性相比特别重要,可以解释物种间的相似性与差异性,这些信息可以更加有效地反映生态型。针对不同环境样品16S rRNA克隆文库之间的微生物群落结构比较已经开发了一些统计技术,如Sørenson、Jaccard、LibShuff 、∫- Libshuff、等级聚类以及基于序列在不同组中的分布进行排序的技术等。由于这些技术没有考虑序列之间相似性的不同程度,所以存在不足之处,但亲缘关系距离的计算可以弥补这些不足。UniFrac是一种基于亲缘关系的β-多样性度量新方法,通过计算包含进化分支的不同群落之间的距离可以同时比较多个样品,其产生的UniFrac 矩阵可以用于多元统计分析[10-13]。这是一款在线分析软件,使用Python程序语言编写,为在微生物生态学中广泛地使用这些技术奠定了基础。而且,这款软件不断改进与完善,并逐步整合一些有效的分析方法。Fast UniFrac是UniFrac的升级版本,基于阵列的实现比基于树的实现使用更少的内存和存储空间,可以使相同的硬件来处理更大的数据集。这些功能的增强使得以前最多只能分析50个样品、5 000条序列、置换次数100次的运行增加到200个样品、100 000条序列、置换次数1 000次的运行。第二代测序技术以及PhyloChip技术使得同时分析数以百计的微生物群落的亲缘关系成为可能。基于缺乏整合和可视化如此庞大数据集的能力,制约了人们对群落结构的了解。Fast UniFrac克服了这些问题,允许在一次分析中整合大量的序列信息和样品信息。新的三维可视化的主坐标分析结果(同时可选择多个坐标轴)提供了一个强大的方法来识别大量的微生物群落的生态类型。升级后的UniFrac既可以用于高通量测序数据的分析,也可以用于PhyloChip技术产生的数据分析[12]。
4 综合数据库在微生物生态学研究中的应用
利用分子生物学技术编目和保护地球上生物多样性的认识在提高、动机在加强,这就需要涵盖三域生命分类系统的综合知识数据库[14-21]。16S小亚基rRNA基因常用于调查微生物群落的组成,推断细菌和古细菌的进化以及设计监测和分析工具,如基因芯片。由于未培养微生物16S小亚基rRNA基因序列的记录产生的速率远超过代表可培养微生物的序列记录,分类放置序列已经相对滞后。这些代表不可培养微生物以及可培养微生物的16S小亚基rRNA序列是通过PCR技术扩增得到,因此存在嵌合体序列,这些低质量的序列会被提交到公共数据库中,如NCBI/EMBL/DDBJ,进而导致微生物分类的相互冲突以及错误采用嵌合体序列推断系统发育或作为参考序列设计探针/引物的概率明显增加[14-21]。要想充分利用这些数据进行可靠的系统发育重建和生物多样性分析,进行每条序列的仔细检查和比对非常必要。特别是最近推出的快速、便宜的测序技术,如焦磷酸测序,进一步证实了需要建立综合的质量控制数据库用于微生物群落的比较。伴随着小亚基(Ribosomal Small Subunit,SSU)和大亚基(Ribosomal Lager Subunit,LSU)rRNA序列的
快速增加,这就需要专门的质量控制数据库和相应的软件工具。目前有4个项目提供了人为策划构建的rRNA序列和比对综合数据库:比利时根特大学建立的欧洲核糖体rRNA数据库,美国密歇根州立大学建立的Ribosomal Database Project II数据库,美国劳伦斯伯克利国家实验室建立的GreenGenes数据库以及德国马克斯·普朗克海洋微生物学研究所建立的Silva数据库[14-21]。4个项目都提供了至少包含16S rRNA基因的数据库,但每个数据库含有的序列数量、质量检查方法、比对方法以及更新程序有所不同。ARB项目是唯一一个整合了同源小亚基(SSU)以及大亚基(LSU)的平台,覆盖了生命的三域分类系统,即细菌域、古生菌域(16S/23S)和真核生物域(18S/28S)的序列。所有项目都提供了基于网络的软件工具用于序列的比对和分类以及探针匹配功能。此外,GreenGenes提供了可与ARB兼容的全长序列(>1 250个碱基)的细菌和古细菌数据集[14-21]。
5 展望
综上所述,微生物生态学研究中,技术进步十分关键,数据分析软件的开发与完善也同等重要,两者缺一不可。
参考文献
[1] SCHLOSS P D,WESTCOTT S L,RYABIN T,et al.Introducing mothur:Open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology,2009,75:7537-7541.
[2] CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods,2010,7(5):335-336.
[3] SHYU C,SOULE T,BENT S J,et al.MiCA:A web-based tool for the analysis of microbial communities based on terminal-restriction fragment length polymorphisms of 16S and 18S rRNA genes[J].Microbial Ecology,2007,53:562-570.
[4] COLLINS R E,ROCAP G.REPK:An analytical web server to select restriction endonucleases for terminal restriction fragment length polymorphism analysis[J].Nucleic Acids Research,2007,35:58-62.
[5] SMITH C J,DANILOWICZ B S,CLEAR A K,et al.T-Align,a web-based tool for comparison of multiple terminal restriction fragment length polymorphism profiles[J].FEMS Microbiology Ecology,2005,54:375-380.
[6] KENT A D,SMITH D J,BENSON B J,et al.Web-based phylogenetic assignment tool for analysis of terminal restriction fragment length polymorphism profiles of microbial communities[J].Applied and Environmental Microbiology,2003,69:6768-6776.
[7] CULMAN S W,BUKOWSKI R,GAUCH H G,et al.T-REX:Software for the processing and analysis of T-RFLP data[J].BMC Bioinformatics,2009,10:171.
[8] SAAL L H,TROEIN C,VALLON-CHRISTERSSON J,et al.BioArray Software Environment (BASE):A platform for comprehensive management and analysis of microarray data[J].Genome Biology,2002,3(8):3.
[9] VALLON-CHRISTERSSON J,NORDBORG N,SVENSSON M,et al.BASE-2nd generation software for microarray data management and analysis[J].BMC Bioinformatics,2009,10:330.
[10] LOZUPONE C,KNIGHT R.UniFrac:A new phylogenetic method for comparing microbial communities[J].Applied and Environmental Microbiology,2005,71:8228-8235.
[11] LOZUPONE C,HAMADY M,KNIGHT R.UniFrac-An online tool for comparing microbial community diversity in a phylogenetic context[J].BMC Bioinformatics,2006,7:371.
[12] HAMADY M,LOZUPONE C,KNIGHT R.Fast UniFrac:Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data[J].The ISME Journal,2010,4:17-27.
[13] LOZUPONE C,LLADSER M E,KNIGHTS D,et al.UniFrac:An effective distance metric for microbial community comparison[J].The ISME Journal,2011,5:169-172.
[14] PhyloTrace:Environmental Sample Analysis[EB/OL].http://www.phylotrac.org/Home.html.
[15] MAIDAK B L,COLE J R,PARKER C T,et al.A new version of the RDP (Ribosomal Database Project)[J].Nucleic Acids Research,1999,27:171-173.
[16] COLE J R,CHAI B,FARRIS R J,et al.The ribosomal database project (RDP-II):Sequences and tools for high-throughput rRNA analysis[J].Nucleic Acids Research,2005,33:295.
[17] COLE J R,WANG Q,CARDENAS E,et al.The Ribosomal Database Project:Improved alignments and new tools for rRNA analysis[J].Nucleic Acids Research,2009,37:141-145.
[18] DESANTIS T Z,HUGENHOLTZ P,LARSEN N,et al.Greengenes,a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J].Applied and Environmental Microbiology,2006,72:5069-5072.
[19] PRUESSE E,QUAST C,KNITTE K,et al.SILVA:A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB[J].Nucleic Acids Research,2007,35:7188-7196.
[20] QUAST C,PRUESSE E,YILMAZ P,et al.The SILVA ribosomal RNA gene database project:Improved data processing and web-based tools[J].Nucleic Acids Research,2013,41:590-596.
[21] SCHLOSS P D.A high-throughput DNA sequence aligner for microbial ecology studies[J].PLoS ONE,2009,4:8230.
猜你喜欢
中国校外教育(下旬)(2016年11期)2016-12-27
中国教育信息化·基础教育(2016年10期)2016-12-20
今传媒(2016年11期)2016-12-19
考试周刊(2016年76期)2016-10-09
