
泡沫镍基电极电解D2O/H2O分离系数测定方法的建立及应用*
2015-12-19 05:28何德良常新园刘芙蓉陈范才
湖南大学学报(自然科学版) 2015年6期
何德良,常新园,熊 英,刘芙蓉,李 菲,陈范才
(湖南大学 化学化工学院,湖南 长沙 410082)
氢同位素的分离与浓缩是核燃料循环、聚变反应堆、聚变 裂变混合堆中必须解决的重要问题之一.到目前为止,虽然国内外学者对其进行了大量的实验与理论研究,已发展出了低温蒸馏、气相色谱、激光分离、薄膜渗透、热扩散等分离方法[1-4].但是由于其难以分离提纯的原因,氘、氚的价格仍很昂贵.电解法是目前国外正在应用的处理含氚废水较为有效的方法之一,电解法浓缩氘高效、节能、环保.尤其近年来发展成熟的SPE(solid polymer electrolyte)电解池具有体积更小、电流密度与电解效率更高、气体纯度更高、使用寿命更长、系统工艺也更简单等优点,而且不会产生放射性废物,获得的气体纯度很高,大大减轻了后续精馏工艺气体纯化的压力.不同的电催化材料电解时氕、氘在电极表面选择性析出的比例不同.其中影响电解水制氢效率的主要因素是由碱性条件下析氢反应(HER)和析氧反应(OER)高过电位而引起的高能耗[5],尤其是析氢电极即阴极电极材料.其中由析氢反应(HER)引起的能耗占相当大的一部分.为了降低反应高能耗问题,在选择合适的析氢反应电极材料时应遵循:1)电极材料必须具有高催化活性.2)电极材料的机械性能、电化学性能稳定.
镍由于具有特殊的d层电子结构,被公认为是具有最高电化学活性的金属[6],在碱性介质中,也具有良好的耐蚀性,因此选用镍及镍合金用作为碱性溶液中析氢反应阴极材料,泡沫镍基合金涂层电极的孔隙尺寸设计为0.3~0.4mm,并具有多层结构.这种特殊的三维多孔结构使其与其他电极相比具有丰富的比表面积,镀层表面丰富,有利于提高氢析出反应(hydrogen evolution reaction,HER)催化性能.但是目前尚缺一种有效评价电极材料分离提纯氕、氘能力的方法,以便能够快速而又有效地寻找合适的电极材料.为此,本文建立一种电解水分离系数的测定方法,并应用于碱性条件下泡沫镍基电极电解重水溶液的分离系数的测定,考察不同电极材料电解水时对氕氘的分离性能.
分离系数的大小反映了电催化材料的分离能力,在仅含有氕氘二种氢同位素的体系中分离系数定义为液相中的氕氘比除以气相中的氘氢比,其表达式为:
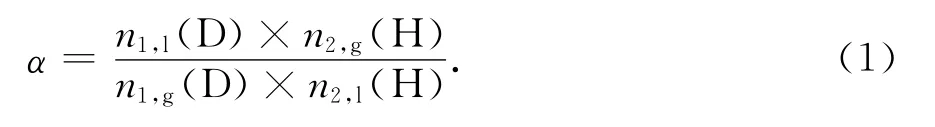
式中:n1,l(D)和n1,g(D)分别为液相和气相中的氘元素的物质的量,n2,l(H)和n2,g(H)分别为液相和气相中氕元素的物质的量,“l”表示液相,“g”表示气相[7].
根据分离系数的公式,对电催化材料进行测试时,必须分别检测出气相中的氕氘比和液相中的氕氘比.M.HAMMERLI和W.J.OLMSTEAD 报道了使用在线质谱法和折射仪法分别检测气相和液相中的氕氘含量[8],该检测系统复杂庞大,不适合普通实验室环境.KRETSCHMER 等的研究报道中液相和气相中的氕氘含量都是通过质谱检测,但是其中检测系统涉及到较复杂的管路连接,不适于在普通实验室建立该体系[9].也有不少文献单独报道了气相和液相中氕氘含量的检测.周俊波研究组进行了低温下气相氕氘的分离及检测[10],但并未涉及到液相中氕氘含量的检测和分离系数的测定.日本学者Yoshinori Kawamura等报道了低温下气相色谱法分析检测氢同位素[11].李桂花和郑彦巍等报道了红外光谱法测定中等浓度重水含量[12].Knež-zeviĉ报道了红外光谱法测试重水浓度[13].这些文献中报道的方法基本上都是针对纯水体系的检测,不适应于电解碱性混合水体系(氢氧化钾的混合水溶液)的测定.
本文在液氮低温冷却下,根据氘气(D2)、氘化氢(HD)和氢气(H2)的气相色谱特征,建立了检测气相中氘气(D2)、氘化氢(HD)和氢气(H2)的工作曲线,根据碱性条件下液态D2O,HDO 和H2O 的红外光谱特征,建立了检测碱性混合水体系的液相中D-O 和H-O 的工作曲线.当对碱性氧化氘/氧化氢混合水溶液进行电解时,可分别测定电解产生的混合气体中的氕氘比(n1,g(D)/n2,g(H))和电解后碱性混合水溶液中的氕氘比(n1,l(D)/n2,l(H)),根据气相中的氕氘比和液相中的氕氘比按式(1)计算得到不同电极材料电解碱性D2O/H2O 混合水溶液时的分离系数.并将该方法应用于测定几种泡沫镍基电极材料电解水时的分离系数.
1 实验部分
1.1 仪器和试剂
电解槽(自制),直流电源,气相色谱(岛津GC-2014C),红外分光光度计(TJ270-30A),散热器,改性泡沫镍基电极,电流表,液氮,液氮罐,氦气(载气),自制氧化铝改性不锈钢填充柱(2根).重水(百灵威,含D 量99.8%),氢氧化钾(AR,国药试剂).
1.2 碱性混合水的红外光谱测试
水中的3种成份H2O,HDO 和D2O 之间能快速建立平衡反应H2O+D2O=2HDO,显然,水中氘原子总含量与3种成份的含量存在关系.红外光谱测试的原理是基于水分子中氢氧键和氘氧键的伸缩振动和弯曲振动而对红外光产生吸收.薄膜法测试水的红外光谱时,薄膜中的每一个氢氧键和氘氧键都会有吸收.无论所形成的薄膜有多薄,该薄膜都是由若干个水分子重叠得到的厚度,那么薄膜中氢氧键和氘氧键的含量与其对红外的吸收强度之间就会存在一定的关系.经本实验证明,在对含中等浓度范围的重水进行红外光谱测试时,存在如式(2)的关系.
取一系列体积的重水V1,l(D2O)和 轻 水V2,l(H2O)配制氢氧化钾质量分数为30%的碱性混合水标准溶液.V1,l(D2O)/[V1,l(D2O)+V2,l(H2O)]值的变化范围为65%~90%.采用液膜法测试了不同重水含量的碱性混合水溶液的红外光谱特征.
1.3 混和气体的气相色谱测试
氢同位素性质尤其是化学性质的差异较小,然而通过已有的实验研究发现:在氢同位素吸收/吸附法分离中,它们之间在一定的实验条件下表现出不同的吸附行为.Katorski等[14-15]介绍了通过变量分离与相关数学近似来求解氢同位素分子在吸附剂上的转动- 振动能以及分离因子的思路,其求解过程较为复杂,得到的理论计算值比实验测得值要高得多.虽然模型需要进一步的修正与完善.但是可以从简化的计算结果中定性地解释和指导氢同位素吸附分离实验中氢同位素气体分子之间的选择性吸附差异与温度、压力及分子零点能之间的依赖关系.同位素气体分子的电子本征能量函数和核间距相同,因此力常数相同.然而由于约化质量不同,所以谐振频率不同.另外,对于轻同位素分子,约化质量μ越小,零点能E0越大;同位素分子越重,振动能级越密集,零点能E0越小,在吸附剂表面容易被吸附.所以吸附剂是优先吸附重的氢同位素气体分子,因为吸附相中较轻的吸附质分子具有较高的量子能态.其次,温度越低,氢同位素气体组份之间的选择性越大,这也是实验中选择液氮温度吸附(65K)的原因.
本文在低温下(65K),以氦气为载气的改性氧化铝所制分离柱能导致氢、氘同分异构体的核自旋发生分离.因为温度在64~77.3K 时,HD/O-H2分离系数一致从而难以分离开.但是氧化铝表面的催化剂使邻 -对位快速互变,从而使沿着分离柱相对运动而引起部分邻 对位同分异构体在一定时间间隔内失去平衡(保留时间)而出现单峰.因为HD 不受影响,进而使H2,HD 和D2得以分离[16].
本文使用二个相同规格和型号的电解槽同时电解纯轻水和含氘量99.8%的重水,分别制取纯净H2和D2配制成一系列的V1,g(D2)/[V1,g(D2)+V2,g(H2)](同温同压下体积之比等于物质的量之比,即n1,g(D2)/[n1,g(D2)+n2,g(H2)])为不同值的标准样品.气相色谱分离检测H2,HD 和D2的基本实验参数:TCD 检测器温度,100℃;最大桥流值,100mA;填充柱和参比柱长,3m;柱温,-196 ℃;柱箱温度,60 ℃;进样口温度,80 ℃;进样量,1mL;载气流速,30mL/min.
1.4 改性泡沫镍基材料的电解分离性能测试
电解试验采用宽8mm,长45mm 的改性泡沫镍作为阴极,同种材料的泡沫镍材料作为阳极,质量分数为30%的氢氧化钾混合水溶液作为电解质.
采用红外光谱法分析电解后碱性混合水溶液的红外光谱特征,并根据液相氕氘比的工作曲线确定液相中的氕氘比.采用低温气相色谱检测气相中D2,HD 和H2的含量,并计算得到气相中的氕氘比.根据电解后气相中的氕氘比和液相中的氕氘比,按式(1)计算不同电极材料的分离系数.
2 结果与讨论
2.1 碱性混合水中氘氢比的工作曲线
利用红外光谱法多次测试所配制的标准溶液,图1所示为65%~90%标准溶液的红外测试图谱.从图中可以看出,氢氧键的最大吸收在3 444cm-1处,将氕氧键在3 444 cm-1处的透光率记为T(OH),氘氧键的最大吸收在2 544cm-1处,氕氧键在2 544cm-1处的透光率记为T(OD).根据DO 键和H-O 键在最大吸收波数处的透过率与它们的含量可以实现液相中氕氘含量地测试.
考虑到氢氧化钾中的氢氧键,并根据V1,l(D2O)和V2,l(H2O)可计算各标准溶液中的D-O 和H-O 之间的含量比.

式中:n1,l(OD)为液相中氘氧键的物质的量;n2,l(OH)为液相中氢氧键的物质的量;n1,l(D2O)为液相中重水的物质的量;n2,l(H2O)为液相中轻水的物质的量;n3,l(KOH)为氢氧化钾的物质的量.
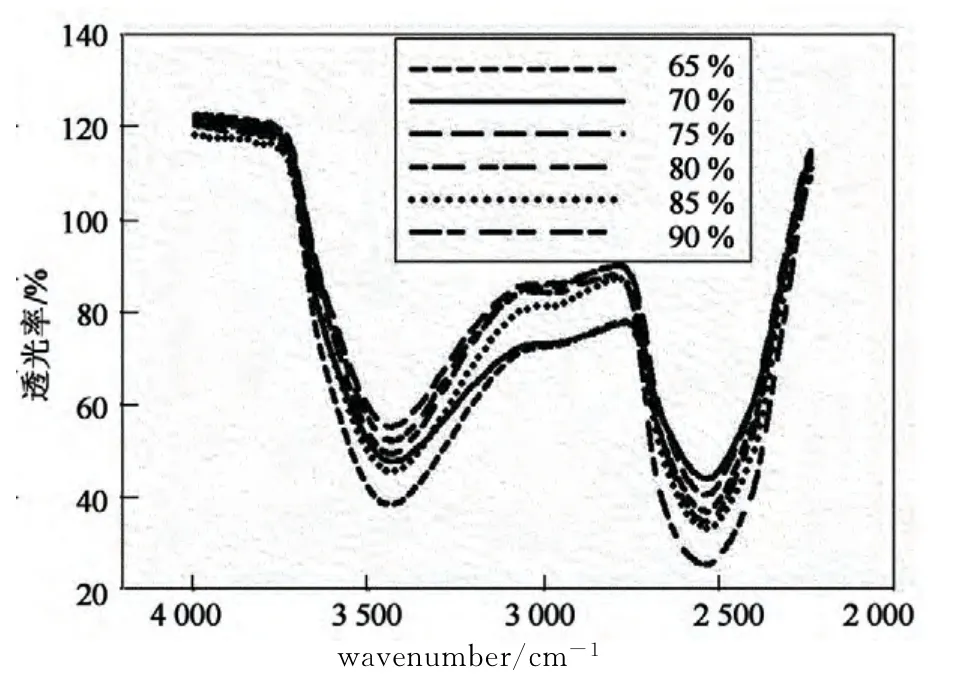
图1 标准溶液的红外测试图Fig.1 The IR curves of standard solution
以T1,l(OD)/[T1,l(OD)+T2,l(OH)]为 横 坐标,以n1,l(OD)/[n1,l(OD)+n2,l(OH)]为纵坐标建立工作曲线(如图2).通过线性回归分析可得液相中的工作曲线为:

式中:X为T1,l(OD)/[T1,l(OD)+T2,l(OH)]值,Y为n1,l(OD)/[n1,l(OD)+n2,l(OH)]值,T1,l(OD)和T2,l(OH)分别为待测溶液中氘氧键和氢氧键红外光最大吸收波长处的透过率.

图2 液相中的测试工作曲线Fig.2 The work line of determination in liquid phase
2.2 气相中各组分的测试工作曲线
图3为V1,g(D2)/[V1.g(D2)+V2,g(H2)](65%~90%)的标准样品色谱测试层叠图.等量的不同物质因热导系数不同而具有不同的响应值,同种物质不同量响应峰面积不同,相同的测试条件下,根据保留时间定性,峰面积定量,从而据此对D2,HD 和H23 种气体物质定量.图3 中保留时间最长的为D2,其次是因H2和D2混合过程中发生交换的极少量的HD,保留时间最短的是H2.
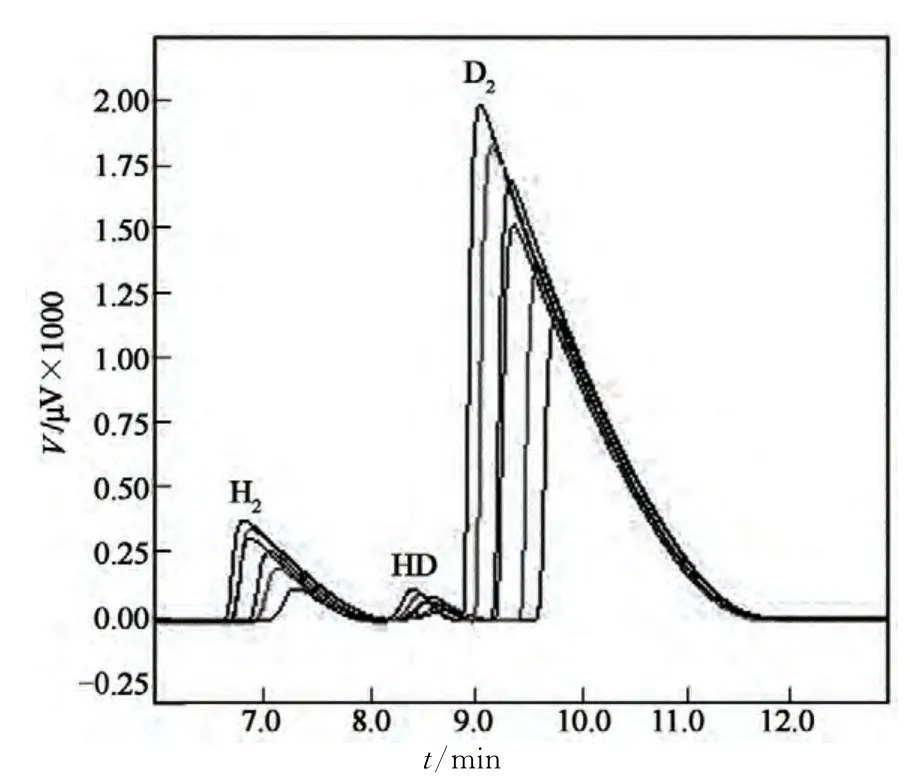
图3 标准样品气相层叠图Fig.3 The Tiered GC picture of standard sample
依据D2测试峰面积与摩尔量n1,g(D2)之间的关系建立并拟合出D2的工作曲线.根据所配标准样品色谱测试数据,进样量均为1mL,以D2响应峰面积为横坐标,物质的量n1,g(D2)为纵坐标建立D2的工作曲线,拟合曲线如图4(a),式中X1为D2响应峰面积,YD2为D2的摩尔量n1,g(D2).曲线方程为:
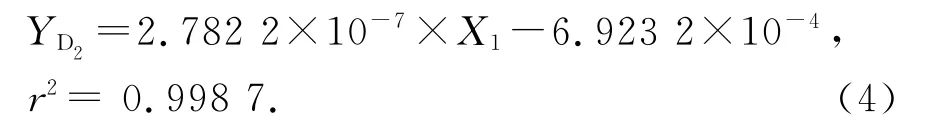
同理依据H2测试峰面积与摩尔量n2,g(H2)之间的关系建立并拟合出H2的工作曲线.以H2响应峰面积为横坐标,物质的量n2,g(H2)为纵坐标建立H2标准曲线,标准曲线如图4(b)所示,式中X2为H2响应峰面积,YH2为H2的摩尔量n2,g(H2).曲线方程为:

HD 标准气体难以配制,而HD 分子是由1 个氢原子和1个氘原子组成的混合分子,它的响应值应处于氢分子和氘分子响应值的中间.为此,HD 标准曲线的建立通过取H2和D2响应峰面积的平均值作为HD 的响应峰面积[9],即在同样浓度下,HD 的响应峰面积=(H2峰面积+D2峰面积)/2.根据D2和H2响应峰面积计算出的HD 的响应峰面积为横坐标,相应的摩尔量为纵坐标作标准曲线,如图4(c),式中X3为HD 响应峰面积,YHD为HD 的摩尔量n3,g(HD).曲线方程为:


图4 气相中各组分的测试工作曲线Fig.4 The work line of ingredients determination in gas phase
2.3 改性泡沫镍基电极材料的氘氢分离系数
试验中所测试的4种电极均为改性的泡沫镍基电极,其编号及成份如下;N-O(wNi=100%),N-F(wNi=82.45%,wFe=17.55%),N-M-C1(wNi=72.45%,wMo=26.02%,wCo=1.53%),N-M-C2(wNi=84.38%,wMo=12.91%,wCo=2.71%).下面以N-F为例进一步说明分离系数的测试和计算.
电解后液相的红外光谱测试结果TN-F(OD)/[TN-F(OD)+TN-F(OH)]为0.528 3±0.007 1,将该值代入式(3)中得到:nN-F,l(OD)/[nN-F,l(OD)+nN-F,l(OH)]为0.520 7±0.010 6,进一步得出nN-F,l(OD)/nN-F,l(OH)为1.086 3±0.045 0,即nN-F,l(D)/nN-F,l(H)为1.086 3±0.045 0.N-F作为电极材料电解所得的混合气体的气相色谱数据:S(D2)为4 155±5,S(H2)为18 535±7,S(HD)为28 111±10,将S(D2),S(H2)和S(HD)分别代入式(4)~(6)中得:nN-F,g(D2),nN-F,g(H2)和nN-F,g(HD)分别为:4.637±0.014×10-4mmol,2.832±0.001×10-2mmol和1.252±0.001×10-2mmol.代入式(7)
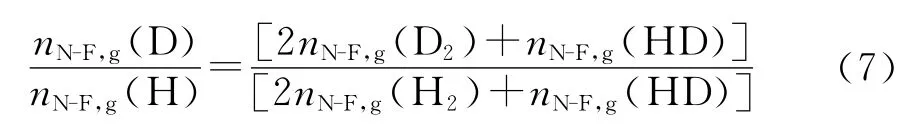
得nN-F,g(D)/nN-F,g(H)为0.194 4±0.000 1.将所得到的nN-F,l(D)/nN-F,l(H)和nN-F,g(D)/nN-F,g(H)值代入式(1)计算出电极材料N-F 的分离系数值为5.6.所有电极材料分离系数测试结果如表1所示.
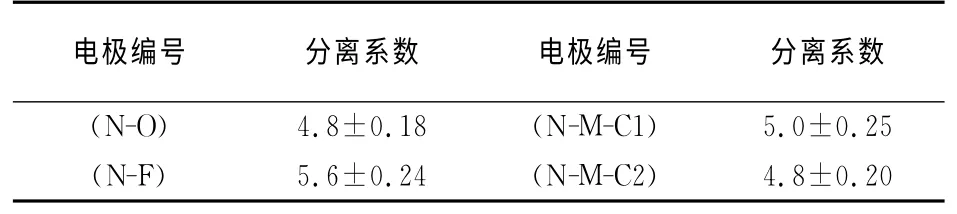
表1 测试电极及其分离系数Tab.1 The determined electrode and their separation factors
为了验证本实验所建立的测试方法,对同一电极材料泡沫镍基电极(N-M-C1)进行了24h连续电解试验并测试其分离系数,每隔45 min测试一次,并绘制出分离系数随时间的变化曲线,如图5所示.有相关文献报道分离系数随电解电流密度的增大而增大,随电解温度的升高而减小.本测试中恒定了电极面积和电解电压,即电流密度恒定,并同时通过冷却水以恒定电解温度.从图中可以看出所测试电极的分离系数值基本与电解时间无关,同时说明了该方法的可靠性.
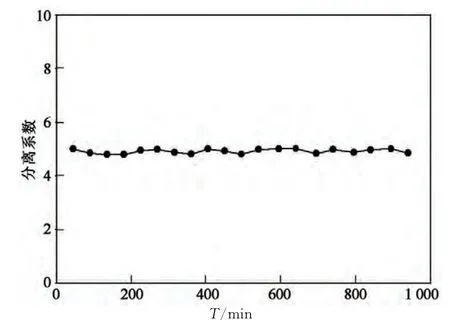
图5 分离系数随时间的变化曲线Fig.5 The variation curve of separation factor with time
3 结 论
建立了一种使用泡沫镍基电极电解碱性混合水溶液并测定其对氘、氢的分离系数的方法.其中,液相中的氘氢比测试使用薄膜法通过红外光谱法定量,制样简单,测试速度快.气相中的氘氢比测试使用廉价的氧化铝经改性后作为填充柱填料,自制参比-分析双柱在液氮冷却下作为分离体系,H2,HD及D2经过分离体系后被TCD 检测器检测,H2,HD和D2分离效果良好,峰型尖锐.
该方法选择性和重现性较好,且简单易行,所需设备廉价.采用本方法所测试计算得到的分离系数与文献报道结果基本一致,且偏差在5%以内.利用本测试方法能对电极材料分离能力做出评价和对比.
[1]孟建波,桑革.SPE 水电解进行H/D 同位素分离研究[C]//中国核学会核材料分会2007年度学术交流会论文集.北京:中国核学会,2007:352-355.
MENG Jian-bo,SANG Ge.H/D isotope separation research in SPE water electrolysis[C]//The 2007Chinese Nuclear Society Conference Proceedings.Beijing:The Chinese Nuclear Society,2007:352-355.(In Chinese)
[2]邓潇君,罗德礼,钱晓静.用于氢同位素分离的置换色谱分离材料的研究进展[J].同位素,2010,23(1):53-58.
DENG Xiao-jun,LUO De-li,QIAN Xiao-jing.Development of separation materials containing palladium for hydrogen isotopes separation[J].Journal of Isotopes,2010,23(1):53-58.(In Chinese)
[3]褚效中,赵宜江,周守勇,等.液氮温度下微孔分子筛5A 与Y分离氢同位素气体的研究[C]//第六届全国化学工程与生物化工年会论文集.长沙:中国化工学会,2010:1-5.
CHU Xiao-zhong,ZHAO Yi-jiang,ZHOU Shou-yong,etal.The research of hydrogen isotopic adsorption on 5Aand Y molecular sieve[C]//The 6th Chemical Industry of Engineering and biological Conference Proceedings.Changsha:Chemical Industry and Engineering Society of China,2010:1-5.(In Chinese)
[4]谢波,刘云怒,翁葵平.色谱柱程序升温脱附法分离氢同位素[J].科技导报.2007,25(20):33-35.
XIE Bo,LIU Yun-nu,WENG Kui-ping.Hydrogen isotope separation using temperature programmed desorption with chromatographic column[J].Science & Technology Review,2007,25(20):33-35.(In Chinese)
[5]HAN Qing,LIU Kui-ren,CHEN Jian-she,etal.A study on the electrodeposited Ni-S alloys as hydrogen evolution reaction cathodes[J].International Journal of Hydrogen Energy,2003,28(11):1207-1212.
[6]韩庆,魏绪钧,刘奎仁.镍合金用作电解水析氢阴极的发展现状[J].中国有色金属学报,2001,11(1):158-162.
HAN Qing,WEI Xu-jun,LIU Kui-ren.Development of nickel alloys as HER cathodes for water electrolysis[J].The Chinese Journal of Nonferrous Metals,2001,11(1),158-162.(In Chinese)
[7]ROY L P.Influence of temperature on the electrolytic separation factor of hydrogen isotopes[J].Canadian Journal of Chemistry,1962,40(7):1452-1460.
[8]HAMMERLI M,OLMSTEAD W J,MUJU B L,etal.Electrolytic H/D separation factors on cathode and diffusion sides of iron membranes in H2SO4[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry,1973,43(1),45-58.
[9]KRETSCHMER J,HEITBAUM J.An e.m.s.study of the cathodic H/D separation factor at gold electrodes in sulfuric acid[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry,1979,97(2):211-218.
[10]周俊波,高丽萍.高纯氘中杂质的低温气相色谱分析[J].原子能科学技术,2007,41(3):356-360.
ZHOU Jun-bo,GAO Li-ping.Analysis Technique of impurity in high purity deuterium by cryogenic gas-chromatography[J].Atomic Energy Science and Technology,2007,41(3):356-360.(In Chinese)
[11]KAWAMURA Y,LWAI Y,YAMANISHI Tetal.Analysis of hydrogen isotopes with a micro gas chromatograph[J].Fusion Engineering and Design,2000(49/50):855-861.
[12]李桂花,郑彦巍.红外光谱法测定中等浓度重水[J].同位素,1993,6(3):168-171.
LI Gui-hua,ZHENG Yan-wei.Determination of the concentrated heavy water by infrared spectrometry[J].Journal of Isotopes,1993,6(3):168-171.(In Chinese)
[13]KNEŽ-ZEVIĈŽV.Determination of the D2O cotent in water by Infrared spectrometry[J].Isotopenpraxis,1965,1:69-71.
[14]KATORSKI A,WHITE D.Theory of adsorption of the isotopic hydrogen molecules at low temperatures[J].The Journal of Chemical Physics,1964,40(11):3183-3194.
[15]WHITE D,LASSETTRE E N.Theory of orotho-para hydrogen separation by adsorption at low temperature isotope separation[J].The Journal of Chemical Physics,1960,32(1):72-83.
[16]MOORE W R,WARD H R.Gas-solid chromatography of H2,HD,and D2.Isotopic separation and heats of adsorption on alumina[J].The Journal of Physical Chemistry,1960,64(6):832-832.
猜你喜欢
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
保鲜与加工(2021年1期)2021-02-06
云南化工(2020年11期)2021-01-14
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
制造技术与机床(2017年12期)2017-02-02
浙江大学学报(工学版)(2016年11期)2016-06-05
现代工业经济和信息化(2016年5期)2016-05-17
