
大鼠少突胶质前体细胞(OPCs) 纯化培养及糖氧剥夺(OGD )模型的建立
2015-12-19 07:10付佩彩黄珊珊唐荣华赵东明
中国组织化学与细胞化学杂志 2015年5期
付佩彩 黄珊珊 唐荣华 赵东明
(华中科技大学同济医学院附属同济医院:1神经内科;2骨科,武汉 430030)
少突胶质前体细胞(OPCs)是近年来发现的神经胶质前体细胞,约占成年中枢神经系统(CNS)中所有胶质细胞的5%-8%。OPCs细胞形态为双极、三级或多级,其特异性标记物为 NG2+A2B5[1]。OPCs持续存在于成年CNS中,为处于早期分化的未成熟细胞,在缺血缺氧等导致的脱髓鞘损伤后,少突胶质前体OPCs活化增殖,迁移至损伤处,分化形成少突胶质细胞(OL),进而形成髓鞘,完成髓鞘再生过程[2,3]。最新的研究证实少突胶质细胞还具有供给神经元能量的功能,在生理状态下,少突胶质细胞能代谢转运乳酸为神经元提供能量支持,帮助神经元的存活[4,5]。因此,少突胶质细胞的病理改变不仅可引起CNS广泛的脱髓鞘病变,而且也极可能参与以神经元轴突缺失和神经性萎缩为特性的神经退行性疾病的病理进程。
本实验采用震摇、差速贴壁法培养高纯度OPCs,并进一步观察其分化为OL能力并建立OGD模型,以期为探讨神经退行性病变发病机制建立进一步试验基础。
材料和方法
1.动物与试剂
实验用SD大鼠乳鼠购自华中科技大同济医学院实验动物中心。胎牛血清、DMEM/HIGH GLUCOSE培养液、N2 supplement(100×)、胰蛋白酶、B27 supplement(50×)、DMEM/F12购自美国GIBCO公司;T3购自invitrogen公司;多聚赖氨酸(polylysine,PLL)、DAPI试剂购自美国 Sigma公司;Rat CNTF、Rat FGF Basic、Human PDGF-AA 购自 Peorotech公司。兔抗NG2购自美国neuromarker公司;鼠抗A2B5购自美国Millipore公司;Edu试剂盒购自上海锐博公司;CY3标记羊抗兔IgG、FITC标记羊抗小鼠IgG购自美国Jackson公司。
2.混合胶质细胞培养
出生3d内SD大鼠乳鼠经75%乙醇消毒后,用眼科剪和眼科镊取脑,在PBS液中充分漂洗;把脑剔除脑膜后,脑组织用眼科剪剪成小块,置于37℃0.125%胰酶中消化10 min;含20%胎牛血清的DMEM/F12培养基终止消化,用巴氏管反复吹打脑组织使其分散成细胞悬液,细胞悬液用200目的筛网过滤;过滤后的悬液置4℃的低温离心机中以800 r/min的速度离心8 min。弃去上清液,用培养基重悬细胞沉淀,接种于用多聚赖氨酸预包被的细胞培养瓶(T75)中;置于普通细胞培养箱(37℃、5%CO2)中培养,培养第3天将培养基更换为含20%胎牛血清的DMEM/高糖培养基,之后每3-4天换液一次;直至混合胶质细胞长满培养瓶底(约10d)。
3.OPCs分离纯化培养及鉴定
待混合胶质细胞长满培养瓶瓶底后,拧紧培养瓶瓶盖,并用封口胶封闭瓶口。将培养瓶置于37℃恒温摇床振摇,设参数为180 r/min,先预振摇2h,然后弃上清,更换新的培养基,继续置于37℃恒温摇床振摇,参数不变,振摇15-18h;后转移振摇后的细胞上清,种于未包被的100 mm培养皿中,置于37℃5%CO2培养箱中,使其静置贴壁1h,吸取上清,此步骤可进一步去除小胶质细胞和星形胶质细胞;收集上清于离心管中离心(800 r/min×8 min),弃去上清,用 OPCs培养基(含 10ng/ml PDGF-AA、10ng/ml FGF Basic、1×N2、1×B27的DMEM/F12培养基)重悬后,种于含有PLL预包被的盖玻片的24孔板及96孔板中。
OPCs隔天换液,培养3d后换用OL分化培养基(含 10ng/ml CNTF、50ng/ml T3、1 × N2、1 × B27 的DMEM/F12培养基)以分化培养OL及进行MTT及EdU检测。
4.OGD干预
OPCs纯化培养3d后,将细胞分为Control组及OGD组进行实验。其中Control组在实验开始时换用新鲜培养液,OGD组换用无糖培养液,OGD在含1%O2,5%CO2,37℃条件下培养 0.5h、1h、2h 及 4h。
5.免疫荧光染色
将24孔板中细胞爬片在干预各时间点吸除原培养基,用PBS漂洗3次,后用预冷的4%多聚甲醛固定30min,吸除固定液,PBS漂洗3次,用含0.2%triton-X100的PBS室温破膜15min,吸除破膜液后PBS漂洗3次,BSA封闭1h后,加入预先用抗体稀释液稀释的对应一抗(anti-NG2、anti-A2B5 和 anti-MBP)4℃封闭过夜。PBS清洗,加入对应二抗(FITC羊抗鼠IgG、CY3羊抗兔IgG)避光条件下室温孵育1h。弃二抗,漂洗后DAPI染核8min,PBS漂洗3次,用50%甘油封片,各爬片在荧光显微镜下观察并拍照。
6.MTT测定细胞活力
震摇纯化后 OPCs离心重悬,调整密度至1×106/ml,每孔加入 200μl至 96孔板中,培养 3d后,按照实验分组设计分别换成相应培养基,每组分别设8个平行孔,在各时间点吸除上清,每孔分别加入 50μL MTT(5mg/mL)继续培养,4h后吸除MTT,每孔加入DMSO 100μl,充分振荡,使紫色结晶物溶解,最后在酶标仪上以490nm波长测取每孔的吸光度值(OD490)。
7.EdU检测细胞增殖
将OPCs调整密度至1×106/m l,接种于含有PLL预包被的盖玻片的24孔板,每孔500μl,培养3d后,按照实验分组设计分别换成相应培养基。在各时间点前2小时加入50μM EdU培养基,分别在各设定时间点漂洗细胞后加入细胞固定液。按EdU试剂盒说明书进行染色,染色成功后照相。
8.统计学处理
实验数据采用SPSS17.0统计学软件进行分析,计量数据用均数±标准差()表示,组间比较采用方差分析。检验水准 α=0.05,以P<0.05认为差异具有统计学意义。
结 果
1.OPCs的鉴定及分化
原代培养的OPCs细胞多为双极性,少数为3极或多极。将OPCs做NG2+A2B5免疫荧光染色,结果显示原代培养的OPCs 95%以上表达OPCs特异性标记物NG2+A2B5(图1A)。进一步分化试验显示,原代培养的OPCs可分化为MBP阳性的OL细胞(图1B)。
2.M TT检测OPCs的活力
与Control组相比,OGD组细胞活力在0.5h无明显改变,在干预1h及2h时细胞活力降低;其中在干预2h时细胞活力降低明显,干预4h时细胞活力降低更加显著(P<0.05)(图2)
3.EdU检测OGD不同时间点OPCs增殖
OGD组干预后1h,2h及4h时EdU阳性率较对照组显著降低(P<0.05),干预0.5h时各组间无统计学差异(图3)。

图1 OPCs特异性鉴定分化:A1为NG2染色阳性,A2为细胞核A2B5染色阳性,A3为A1与A2及DAPI的合成图;B1为MBP染色阳性,B2为细胞核DAPI染色,B3为B1与B2的合成图。Bar=25μmFig.1 The specific identification of OPCs and OL:A1:NG2 positive staining,A2:A2B5 positive nuclear staining,A3:DAPIstainingmerged with A1 and A2;B1:MBP positive staining,B2:DAPIstaining,B3:B1 and B2 merged figure

图2 不同干预组OPCs的MTT检测统计图.*P<0.05Fig.2 The statistical figure of MTT test of OPCs by different intervention.*P<0.05
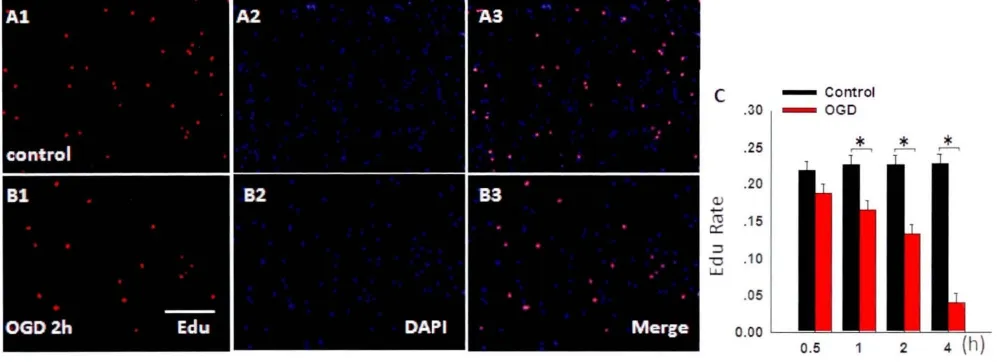
图3 不同干预组EdU阳性率检测结果 A为Control组EdU阳性率(A1为EdU着色,A2为DAPI,A3为合成图);B为OGD2h组EdU阳性率(B1为EdU着色,B2为DAPI,B3为合成图);C各组EdU阳性细胞率的统计图(*P<0.05)Fig.3 Results of EdU positive staining rate in different groups A:EdU positive staining in the Control group(A1:EdU staining,A2:DAPIstaining,A3:A1 and A2 merged);B:EdU positive staining in 2h OGD group(B1:EdU staining,B2:DAPIstaining,B3:A1 and A2 merged;C:The statistical figure of EdU positive rate by different intervention(*P<0.05)
讨 论
颅脑缺血性损伤(卒中)、脊髓损伤、脑室周围白质软化以及多发性硬化等多种CNS疾病均可导致OL大量死亡,引发广泛的脱髓鞘病变,造成神经功能损伤[6-8]。目前诸多研究已证实少突胶质细胞功能失调参与了许多神经退行性疾病的致病,以肌萎缩侧索硬化(ALS)为代表的遗传性运动神经元退变疾病中,少突胶质细胞的形态异常改变远早于疾病的发生并且疾病的发展最终导致少突胶质细胞的死亡[9]。成熟的OL自身不可再生修复,其再生依赖于OPCs的增殖分化[10]。OPCs在鼠类胚胎期E 14.5d出现,在成熟中枢神系统中持续存在,在脱髓鞘损伤后,OPCs可迁移并分化为成熟OL,促进髓鞘修复。因此,OPCs的体外培养对研究脱髓鞘疾病及神经退行性病变具有重要价值。
目前报道的OPCs培养方法包括切片培养法、免疫抗体包被筛选法、振摇法、神经球诱导分化法等[11-13]。上述方法像切片培养法主要用于原位研究OPCs细胞行为学,不是以扩增和纯化为目的的培养;免疫抗体包被筛选法耗费抗体多,实验成本大;而神经球诱导分化法容易出现星形胶质细胞及神经元等杂细胞。本实验采用改良振摇法可避免上述耗费成本高等缺陷,且一次培养混合细胞可振摇后重新培养,可重复振摇2-3次,细胞纯度达到95%以上,且可同时得到纯度较高的小胶质细胞,可用于需要大量细胞的试验。然而,在分离培养OPCs过程中,有下列几点需要注意:(1)取材需把标本置于冰上进行,这样可最大限度保持细胞的活性。(2)控制好胰酶消化的时间,限定在10min左右。(3)振摇法的过程设计的选择是根据细胞贴壁牢固程度确定的,悬液中各种细胞贴壁牢固程度从大到小排序为星形胶质细胞、少突胶质前体细胞、小胶质细胞。预摇2h首先可将部分小胶质细胞去除,振摇15-18h后OPCs可从贴壁的混合胶质细胞层摇下,此时收集细胞上清,再行贴壁培养1h,然后取细胞上清,此步骤可进一步去除残留的星形胶质细胞和小胶质细胞,这是因为三种细胞中,OPCs的贴壁时间最长。(4)振摇时封闭培养瓶瓶口的目的是创造缺氧环境,该环境有利于OPCs分离进入悬液中。(5)振摇和贴壁要严格把握参数和时间,否则影响OPCs的纯度和获得细胞量。
近年来,神经细胞体外缺氧培养的同时合并培养基缺糖已被公认为模拟体内氧化应激损伤合适的模型。关于OPCs缺血缺氧损伤的体外试验较少,文献中OGD干预时间及氧浓度不等,其中干预时间主要为 0.5 -2h[14-16]。本试验中,我们通过对 OPCs 糖氧剥夺后,使用MTT检测细胞活力以及EdU试剂盒检测细胞增殖。结果显示,OGD干预0.5h后,与对照组相比,细胞活力及细胞增殖均无明显差别。干预1h、2h及4h后,与对照组相比,OPCs细胞活力及细胞增殖均有下降(P<0.05),其中干预2h时,MTT下降明显,细胞增殖减低约为40-50%,故我们考虑2h可作为OPCs体外糖氧剥夺模型合适的试验损伤时间,干预4h时,细胞损伤明显,故不再延长干预时间。
综上所述,本文中我们采用震摇、差速贴壁法培养出高纯度的OPCs,且培养的OPCs具有分化成为OL的能力。2h可作为OPCs体外OGD模型建立的合适时间。
[1]McCarthy KD,de Vellis J.Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue.J Cell Biol,1980,85(3):890-902
[2]Vitry S,Avellana-Adalid V,Lachapelle F,et al.Migration and multipotentiality of PSA-NCAM+ neural precursors transplanted in the developing brain.Mol Cell Neurosci,2001,17(6):983-1000
[3]Cai J1,Qi Y,Hu X,et al.,Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independentof Nkx6 regulation and Shh signaling.Neuron,2005,45(1):41-53
[4] van der Burg JM,Björkqvist M,Brundin P.Beyond the brain:widespread pathology in Huntington’s disease.Lancet Neurol,2009,8(8):765-774
[5]Bradford J,Shin JY,Roberts M,et al.Expression ofmutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms.Proc Natl Acad Sci U SA,2009,106(52):22480-22485
[6]Kessaris N,Fogarty M,Iannarelli P,et al.Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage.Nat Neurosci,2006,9(2):173-179
[7] Miller,R.H.Regulation of oligodendrocyte development in the vertebrate CNS.Prog Neurobiol,2002,67(6):451-467
[8]Noble M,Fok-Seang J,Wolswijk G,et al.,Development and regeneration in the central nervous system.Philos Trans R Soc Lond B Biol Sci,1990,327(1239):127-143
[9] Philips T,Bento-Abreu A,Nonneman A.Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis.Brain,2013,136(Pt2):471-482
[10]Labombarda F,González S,Lima A,et al.Progesterone attenuates astro- and microgliosis and enhances oligodendrocyte differentiation following spinal cord injury.Exp Neurol,2011,231(1):135-146
[11]Chen Y,Balasubramaniyan V,Peng J,et al.Isolation and culture of rat and mouse oligodendrocyte precursor cells.Nat Protoc,2007,2(5):1044-1051
[12]Hu J,Deng L,Wang X,et al.Effects of extracellularmatrix molecules on the growth properties of oligodendrocyte progenitor cells in vitro.J Neurosci Res,2009,87(13):2854-2862
[13]Tsai HH1,Macklin WB,Miller RH.Distinctmodes ofmigration position oligodendrocyte precursors for localized cell division in the developing spinal cord.J Neurosci Res,2009,87(15):3320-3330
[14]Fu P,Tang R1,Yu Z,et al.Bumetanide-induced NKCC1 inhibition attenuates oxygen-glucose deprivation-induced decrease in proliferative activity and cell cycle progression arrest in cultured OPCs via p-38 MAPKs.Brain Res,2015,1613:110-119
[15]Wu X,Qu X,Zhang Q,et al.Quercetin promotes proliferation and differentiation of oligodendrocyte precursor cells after oxygen/glucose deprivation-induced injury.Cell Mol Neurobiol,2014,34(3):463-471
[16]Wang XQ,Yao RQ,Liu X,et al.Quercetin protects oligodendrocyte precursor cells from oxygen/glucose deprivation injury in vitro via the activation of the PI3K/Akt signaling pathway.Brain Res Bull,2011,86(3-4):277-284
猜你喜欢
热力发电(2022年3期)2022-03-25
数学物理学报(2021年6期)2021-12-21
能源工程(2021年2期)2021-07-21
安徽医药(2020年1期)2020-12-23
医学新知(2019年4期)2020-01-02
中国社区医师(2019年12期)2019-08-26
中国乳品工业(2018年10期)2018-11-16
大众健康(2017年1期)2017-04-13
中国医科大学学报(2016年11期)2016-12-01
上海故事(2015年13期)2016-01-22
