
心房结构重构在心房颤动的发生及维持中的作用
2015-12-16 05:59:23易茜综述马瑞彦审校
中国循环杂志 2015年8期
易茜综述, 马瑞彦审校
心房结构重构在心房颤动的发生及维持中的作用
易茜综述, 马瑞彦审校
心房颤动(房颤)在临床上较为常见,具有致残率和致死率高等特征,但其确切分子机制仍不明确。近年研究表明,心房重构是房颤发生、发展和维持的关键环节,而结构重构在其中的作用更为显著。 本文综述了肾素—血管紧张素—醛固酮系统、基质金属蛋白酶系统、氧化应激、MicroRNA、内皮细胞间质转化(EndMT)、遗传基因多态性等参与结构重构的可能机制,为进一步探讨房颤的发生和临床治疗提供了新的思路。
心房颤动; 心房重构; 结构重构;纤维化;分子机制
心房颤动(房颤)在临床上较为常见,其发病率随年龄增长而不断上升[1],60岁以上人群发病率高达6% 以上[2],随着全社会老龄人口的增多,房颤有向全球性疾病的趋势发展[3]。其主要危害是脑卒中和肢体栓塞等并发症,已成为心血管疾病发病和死亡的主要原因之一,对于房颤的治疗、发病机制和临床治疗策略研究一直是心血管领域的重点。目前,房颤的治疗主要包括内科抗血栓聚集、转复窦性心律、减慢心室率等方面的药物治疗和射频消融术,以及外科的改良迷宫手术(maze),但都存在很大的局限性,包括其治疗的有效性以及术后出现的恶性心律失常、复发率高等[4],其根本原因是房颤的发生机制研究不透彻。故对房颤潜在分子机制的深入研究是提高临床疗效的前提。既往,对房颤的研究主要集中于电生理方面。近年来的研究表明,心肌胶原纤维的含量和构型改变是造成心脏重构的原因,结构重构在房颤中的作用和意义已被广泛关注,本文就近年来关于结构重构的最新进展进行综述。
1 房颤发生的基本机制
经典理论认为房颤的发生是心房内的异位点起搏以及微折返环的形成,多个异位灶相互碰撞形成折返,构成一个环路[5],这两个基本机制目前已得到共识。房颤发生时,心房肌细胞快速激活,通过电生理重构,诱发快速性房性心律失常,心房内多地点的刺激,能够产生空间的异质性,导致结构重构。研究表明,诱发房颤的始动因素和维持其病理状态的基质是房颤相关机制的两个主要部分[6]。目前认为房颤的发生和维持是多因素多通道共同作用的结果。
2 心房电重构
1995年Wijffels首次提出了“心房电重构”这一概念,通过建立快速心房起搏诱发山羊房颤模型发现:反复刺激导致心房有效不应期(AERP)缩短,与刺激时间成反比。刺激时间越长,房颤的诱发率越高,即是”房颤致房颤”理论。研究表明,快速房性心律失常可影响有效不应期的空间分布,增加空间异质性[7],成为触发房颤的始动因素。其机制可能源于钠、钾通道的表达或功能异常,引起心肌电信号传导紊乱,从而诱发心律失常。临床证据也表明: 阵发性房颤患者随着病程的延长,若不加以干预和控制,可发展为持续性房颤。近年来,人们认识到心房的结构重构和电生理重构是房颤发生和维持的重要基础[8],尽管两者在促房颤过程中的作用和意义不尽相同。
3 心房结构重构
Everett等[9]在犬的房颤模型上发现慢性房颤转复为窦性心律后1~2周电生理改变完全恢复, 但心房的细胞形态和收缩功能却不能同步恢复,这说明了影响房颤发生及维持的因素绝不是只有电重构。目前认为结构重构也可致局部心肌电活动传导不均一,形成较多的微折返环,造成传导阻滞;还可影响细胞间连接如缝隙连接蛋白(Cx)数量的缺失以及分布的改变[10],造成病变心脏的不协调收缩,进一步促进房颤的发展。
3.1 心房结构重构时心房肌细胞超微结构改变
心房结构重构主要表现在心肌细胞超微结构的改变、心肌间质胶原纤维重分布及纤维化等。光镜和电镜下可见:房颤动物模型和临床标本90%以上心房肌细胞结构改变, 包括心房肌细胞肥大、溶解、肌纤维丢失、小而异形的线粒体数目增加、肌浆网的断裂、细胞外基质增多、核周糖原堆积、α-肌球蛋白重链(MHC)向β-肌球蛋白重链转化、α-平滑肌肌动蛋白(α-SMA)等胎儿期特征蛋白重新表达,称为“反去分化( dedifferentiation)”; 心房肌加速合成的β-MHC 是胚胎型异构蛋白, 寿命短, 易衰竭, 加速了心肌重塑[11]心房肌结构的改变也给房颤的持续发生提供了一个良好的契机[12]。
3.2 参与心房结构重构的相关分子机制
房颤结构重构是心房纤颤的中心环节,心房纤维化是其主要表现,与其相关的分子机制较多,且各因素间的相互关联错综复杂[13,14],图1简要概括了一些促房颤发生的因素,可望在分子水平揭示房颤本身的发病特点。
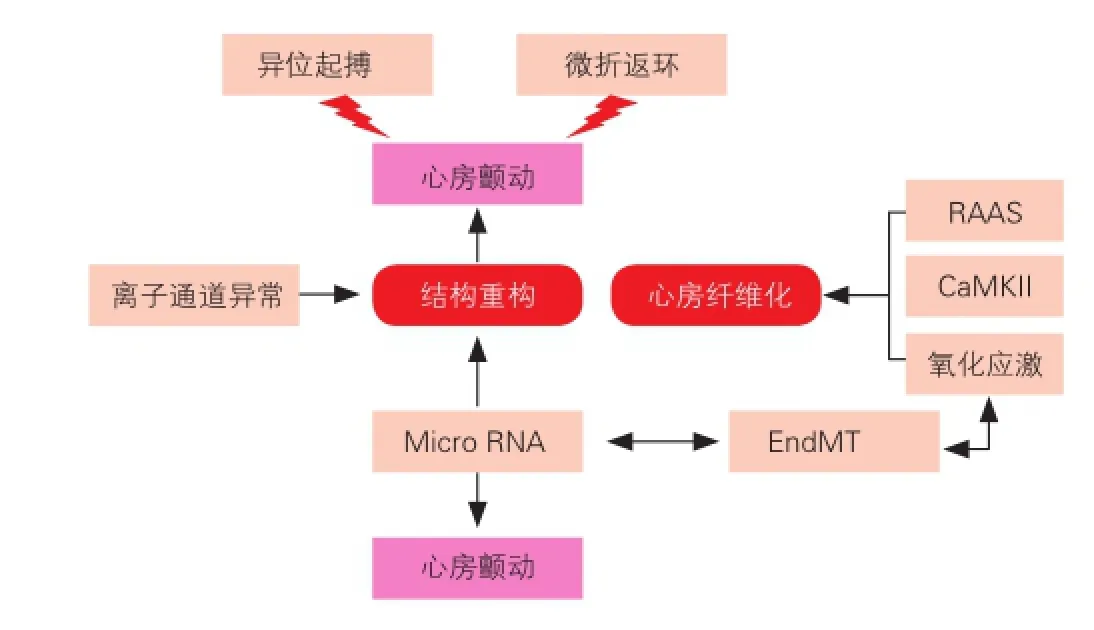
图1 促心房颤动发生的因素
3.2.1 肾素—血管紧张素—醛固酮系统(RAAS)
RAAS系统在肾脏中的作用早已被广泛研究,但对心房重构方面的影响在近几年才开始被广泛关注。房颤发生时,血管紧张素Ⅱ( AngII)生成增加,AngII可诱发心房纤维化,出现传导阻滞、折返易于发生,而关于其致纤维化的机制,有学者认为其通过促分裂原激活蛋白激酶途径诱发心肌间质细胞纤维网络激活而导致心肌间质纤维化,也有研究认为[15],AngⅡ通过增强L-type 钙通道的α1 C亚单位的微孔的表达来增强L-type钙离子电流和转运。另外AngⅡ可以增加心房的压力,并通过增大心房内径和改变心房的电生理重构而引发房颤。AngⅡ还能通过血管紧张素转化酶1受体(AT-1)和AT-2 直接引起心房肌细胞凋亡,AngⅡ与心肌细胞共同孵育24 h能使凋亡细胞百分率较正常对照组增加5倍以上[16],可见AngII能通过多种途径参与心房纤维化。近年来,醛固酮受体(MR)致心肌纤维化作用也日益受到重视[17],临床上也早已有MR拮抗剂作为对抗纤维化的治疗,代表性药物如我们熟悉的螺内酯(安体舒通),在小鼠动物模型上已验证增长的醛固酮水平能促进心房纤维化的进展,但有效性仍有待进一步研究。
3.2.2 基质金属蛋白酶系统(MMPs)
MMPs 是一类降解胶原的最主要蛋白水解酶系统,它一方面介导胶原合成,促进间质纤维化,另一方面可促进胞间胶原分解断裂,从而使心房结构疏松,发生间质重构,并主要以降解为主。MMPs与金属蛋白酶组织抑制因子(TIMPs) 的平衡决定了细胞外基质( ECM)的含量[18]。新近研究证实房颤时心房肌MMPs表达增加,使正常的ECM 成分降解, 合成异常的胶原蛋白及结缔组织, 参与心房结构重构。MMP-2、MMP-9是目前研究较多的明胶酶族[19]。Huxley等[20]发现房颤患者心房组织中MMP-9的活性升高, 且其活性与左心房直径呈正相关, 表明MMP-9与房颤过程中的心房扩张及心房结构重构有相关性。Xu等[21]观察心脏移植患者发现, 伴房颤者的心房肌较窦性心律者I型胶原容积分数明显增加, 左心房增大,且MMP-2、MMP-9活性增高, 永久性房颤者更为突出;房颤患者TIMP-2蛋白水平下调;且心房MMP-2/TIMP-2比值与I型胶原容积分数呈正相关。上述研究结果初步提示, MMPs活性增高和TIMPs表达水平降低可能在房颤心房扩张和心肌重塑中起到了一定的作用, 二者之间的动态失衡会加速心肌重塑的进程。
3.2.3 炎症和氧化应激(OS)
炎症过程包括组织损伤、促炎因子释放、炎症细胞渗出、新生血管形成和瘢痕形成等环节。近年很多研究表明炎症因子与许多器官(心、肺、肾、皮肤等)的纤维化联系紧密[22,23]。氧化应激是指病理状态下机体促氧化与抗氧化失衡时, 并产生过量活性氧自由基(ROS), 直接引起心肌细胞损伤和坏死, 导致心肌间质纤维化。ROS的促纤维化作用包括促成纤维细胞增加、向基质分生的肌原纤维转变、促进前纤维基因表达的上调及引起MMPs/TIMPs失衡等。Lin等[24]采用荧光定量聚合酶链式反应(PCR)技术对14例慢性房颤患者右心耳线粒体研究发现,房颤患者线粒体DNA(mtDNA)氧化损伤程度较窦性心律者重, mtDNA氧化损伤最常见产物之一8-羟基-2脱氧鸟嘌呤核苷以及mtDNA含量较窦性心律者显著增加, 故认为房颤患者心房肌 mtDNA存在氧化损伤。动物模型研究提示氧化应激相关蛋白可预测房颤发生。目前,ROS的促纤维化作用已被广泛接受,但是其来源仍有争议。研究者通过对烟酰胺腺嘌呤二核苷酸(NADPH)氧化酶缺乏的小鼠研究发现,其发生氧化应激的机率小于正常对照组,故认为NADPH氧化酶可能作为ROS来源参与了结构重构。
近来有关房颤的大量研究均提示多功能的钙调素依赖蛋白激酶Ⅱ(CaMKII)能作为ROS的感受器,是发生心律失常前的一种信号[25],发生氧化应激时表达为ox-CaMKII,感受Ca2+浓度变化,增加了房颤的易感性。Purohit等[26]发现:注射了AngII的小鼠在经过快速心房刺激后房颤的易感性较对照组小鼠明显增强。在临床工作中使用药物干预CaMKII转化为ox-CaMKII可能作为一种新的治疗靶点。
3.2.4 MicroRNA
MicroRNA(miRNA)是近来发现的一类高度保守、非编码的、能在翻译水平调节mRNA表达的小分子RNA。而最新研究发现miRNA 与心肌纤维化关系密切。miRNAs 能直接参与心肌纤维化,作用于心脏成纤维细胞,调控心脏成纤维细胞的存活[27],这些miRNAs在不同环节参与心房的结构重构,从而在房颤的发生与维持中发挥重要作用[28]。MicroRNA-29a 和-133a 在心肌中特异性表达与纤维化密切相关[29,30],Duisters等[31]研究发现miR-133过表达可使结缔组织生长因子mRNA表达水平下降,减少心肌胶原蛋白的生成,抑制心肌纤维化,而且miR-133在人和鼠的胚胎、成人心脏及骨骼肌中均有表达,其表达水平和心肌肥厚存在负性相关。miR-29 表达于成纤维细胞中,调控mRNAs以编码多种纤维蛋白、胶原蛋白、弹性蛋白等与心肌纤维化相关的蛋白。有研究者从mircroRNA 角度提出类似肿瘤发生学说中的“平衡”学说,即正常情况下,心脏中数百的microRNA 在调控靶基因时处于一种平衡状态,而当某些因子介入后,打破了平衡,致使其调控的靶基因表达发生改变,导致心脏电重构及结构重构的发生,从而诱发房颤。
3.2.5 内皮细胞向间充质细胞的转分化(EndMT)
EndMT是指病理或生理状态下,内皮细胞在受到刺激因子的作用下,发生形态、结构和功能上的改变,具体表现为:内皮细胞的主要标志物血小板内皮细胞黏附分子(PECAM-1/ CD31)等表达减弱或消失,表达为成纤维细胞标志物如α-平滑肌肌动蛋白(α-SMA)、波形蛋白(vimentin)、成纤维细胞特异性蛋白-1(FSP-1,也被称作S100A4)、I型和III型胶原蛋白等。有研究发现:小鼠心肌纤维化过程中有EndMT参与,并推测出约27%~35%的成纤维细胞来源于经EndMT转化的内皮细胞,心肌纤维化程度减轻的同时常伴有EndMT减少[32]。体外培养人真皮层的微血管内皮细胞,给予炎症因子刺激,能表现出成纤维细胞特性,表达出α-SMA、钙调蛋白和Ⅰ型胶原蛋白等,提示真皮微血管内皮细胞在炎症因子诱导下发生了EndMT[33]。Ghosh等[34]利用miRNA芯片技术检测了转运生长因子(TGF)-β诱导的内皮源性(发生了EndMT)成纤维细胞内miRNA水平,发现miRNA-125b、miRNA-21和miRNA-30b水平在EndMT过程中显著升高,相反另外一些miRNA,如miRNA-122a、miRNA-127明显降低。近年,对miRNA调节EndMT机制的研究已成为一大热点,并取得了不少成果,可能为抑制病理状态下EndMT,进而为抗心肌纤维化找到新的靶点。
3.2.6 遗传基因多态性
人类基因组存在着约140多万单核苷酸多态性(SNP),Ng报道人的基因组约有3000多个SNP,其中1/4是有害的,肾上腺素能受体(β2-AR)是心血管上重要的受体亚型,其基因多态性与高血压、心力衰竭、心肌肥大、肥胖、血脂等病变密切相关。众多研究也证明ACE两种等位基因( I/D)多态性与左心室扩张及扩张性心肌病密切相关。 ACE I/D多态性决定了体内的ACE水平,释放大量的RAAS,促进心房纤维化,参与结构重构[35]。心血管系统往往涉及涉及多个基因以及环境的相互作用,尤其大多数心血管疾病具有遗传复杂性,根据基因型决定个性化治疗方案是未来的一种理想趋势,但目前因研究局限,仍有大量工作需要完成。
3.3 心房逆重构
近来,心房逆重构越来越得到重视,为预防和治疗房颤提供了最新思路。逆重构(reverse remodeling)是指房颤转复后心脏电生理特征及收缩功能得以恢复,组织结构去分化程度减轻。逆重构是个缓慢过程,尤以结构重构的逆转更甚。Ausma等[36]研究房颤转复后心肌溶解、肌浆网断裂、心肌间连接组织等去分化改变逆转,发现转复后收缩功能恢复缓慢,可能是持续性房颤加剧了心房肌细胞的牵张,增加了Ca2+超载,心肌组织结构改变,延缓转复后收缩重构的逆转。另有研究证明,慢性AF患者在经过导管射频消融术后,电重构恢复较快,心室率得到控制,但心脏结构重构作用并未停止,房室不同步及不规则的心室率可能在AF患者心脏结构重构中发挥重要作用[37]。Everett[9]等的动物模型研究发现,房颤转复后4h即可观察到逆重构改变,7~14天后心房有效不应期和房颤周期长度可完全恢复,但未观察到结构重构恢复的改变。ACEI和ARB类药物对于逆转心脏重构有重要作用,其确切机制不明,可通过降低心房内压力,抑制心房纤维化,改善心房内延迟传导等,相关的RAS系统抑制剂药物试验仍在进行中。充分研究心房逆重构的过程和机制对于找到房颤治疗的新靶点带来了新的思路。
4 展望
综上,房颤是一种复杂的心律失常,有多种调节机制及细胞内信号转导途径共同参与,各种机制间又存在交叉效应或共同通路,由于参与心房结构重构的因素和途径的复杂性,可能对治疗带来一定难度,但阻止及逆转心房结构重构可能成为未来治疗房颤的新策略。虽然在多数患者中我们仍不能确定房颤的发生发展机制,在今后的基础及临床研究中,可能需要从更多角度开展研究以进一步了解房颤的发生发展机制,并进行相关的临床干预以验证治疗效果。对心房结构重构相关分子机制的深入了解,可为房颤的临床治疗提供新的思路。
[1] Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation, 2006, 114: 119-125.
[2] Singh D, Cingolani E, Diamond GA, et al. Dronedarone for Atrial Fibrillation: Have we expanded the antiarrhythmic armamentarium . J Am Coll Cardiol, 2010, 55: 1569-1570.
[3] Wilke T, Groth A, Mueller S, et al. Incidence and prevalence of atrial fi-brillation: an analysis based on 8. 3 million patients . Europace, 2013, 15: 486-493.
[4] Nattel S. From guidelines to bench: implications of unresolved clinical issues for basic investigations of atrial fibrillation mechanisms. Can J Cardiol, 2011, 27: 19-26.
[5] 马瑞彦, 肖颖彬. 心房颤动致心房重构的分子机制研究进展. 中国循环杂志, 2005, 4: 313-316.
[6] Miyagawa S, Sakaguchi T, Nishi H, et al. Recent clinical and experimental advances in atrial fibrillation. ISRN Cardiol, 2011, 2011: 1-12.
[7] Park JH, Pak HN, Lee S, et al. The clinical significance of the atrial subendocardial smooth muscle layer and cardiac myofibroblasts in human atrial tissue with valvular atrial fibrillation. Cardiovasc Pathol, 2013, 22: 58-64.
[8] Xu Y, Sharma D, Li G, et al. Atrial remodeling: New pathophysiological mechanism of atrial fibrillation. Med Hypotheses, 2013, 80: 53- 56.
[9] Everett TH , Li H , Mangrum JM , et al . Electrical morphological , and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation . Circulation , 2000 , 102: 1454-1460.
[10] Igarashi T, Finet JE, Takeuchi A, et al. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation , 2012, 125: 216-225.
[11] 阮红梅, 王玮. 心房颤动发生机制的研究进展. 医学综述, 2011, 17: 37-40.
[12] Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol, 2008, 51: 802-809.
[13] Nattel S, Guasch E, Savelieva I. Early management of atrial fibrillation to prevent cardiovascular complications. Euro Heart J, 2014, 35: 1448-1456.
[14] Harada M1, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J, 2015, 79: 495-502.
[15] Tsai CT, Wang DL, Chen WP, et al. Angiotensin II increases expression of alpha1C subunit of L-type calcium channel through a reactive oxygen species and cAMP response element binding proteindependent pathway in HL-1 myocytes . Circ Res, 2007, 100 : 1476-1485.
[16] Von Lewinski D, Kockskamper J, Rubertus SU, et al. Direct proarrhythmogenic effects of angiotensin Ⅱ can be suppressed by AT1receptor blockade in human atrial myocardium . Eur J Heart Fail, 2008, 10: 1172-1176.
[17] Young MJ. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr Opin Nephrol Hypertens, 2008, 17: 174-180.
[18] Kim SK, Park JH, Kim JY, et al.High plasma concentrations of transforming growth factor-β and tissue injibitor of metalloproteinase-1--potential non-Invasive predictors for electroanatomical remodeling of atrium in patients with non-valvular atrial fibrillation. Circ J, 2011, 75: 557-564.
[19] 董丽君, 汤宝鹏, 许国军, 等. 心房肌基质金属蛋白酶及其抑制剂、凋亡相关基因表达改变与增龄性心房颤动关系的研究. 中国循环杂志, 2012, 29: 1034-1038.
[20] Huxley RR, Lopez FL, MacLehose RF, et al. Novel Association between Plasma Matrix Metalloproteinase-9 and Risk of Incident Atrial Fibrillation in a Case-Cohort Study: The Atherosclerosis Risk in Communities Study. PLOS one, 2013, 8: 1-8.
[21] Xu J, Cui G, Esmailian F, et al. Atrial extracellular matrix remodeling and the main- tenance of atrial fibrillation . Circulation, 2004, 109: 363.
[22] Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol, 2010, 43: 161-172.
[23] Zeisberg EM, Potenta SE, Sugimoto H, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to- mesenchymal transition. Am Soc Nephrol, 2008, 19: 2282-2287.
[24] Lin PH, Lee SH, Su CH, et al. Oxidative damage tomitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic BiolMed, 2003, 35 : 1310.
[25] Erickson JR, He BJ, Grumbach IM, et al. CaMKII in the Cardiovascular System: Sensing Redox States. NIH Public Access, 2011, 91: 889-915. [26] Purohit A, Rokita AG, Guan X, et al. Oxidized Ca2+/calmodulindependent protein kinase II triggers atrial fibrillation. Circulation, 2013, 128: 1748-1757.
[27] Lu Y, Zhang Y, Yang B, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation, 2010, 122: 2378-2387.
[28] 何英杰, 郭玲, 丁振华. miRNA分子miR-24的研究进展. 解放军医学杂志, 2009, 34: 1145-1147.
[29] Chen S, Puthanveetil P, Feng B, et al.Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. Cell Mol Med, 2014, 18: 415-421.
[30] He Y, Huang C, Lin X, et al, Micro R NA-29 family, a crucial therapeutic target for fibrosis diseases . Biochimie, 2013, 95 : 1355-1359.
[31] Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res, 2009, 104: 170-178.
[32] Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-tomesenchymal transition contributes to cardiac fibrosis. Nat Med, 2007, 13: 952-961.
[33] 舒晓蓉, 聂如琼, 谢双伦. 内皮间质转化和心肌纤维化. 中华心血管病杂志, 2014, 42: 797-800.
[34] Ghosh AK, Nagpal V, Covington JW, et al. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal, 2012, 24: 1031-1036.
[35] 张玲, 桂庆军, 彭建业. ACE基因多态性与心血管疾病相关性的研究进展. 中国心血管病研究, 2010, 8: 460-462.
[36] Ausma J, van der Velden HM, Allessie MA. Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation. 2003, 107: 2051-2058.
[37] 刘飞, 徐建, 严激. 射频消融术对心肌病阵发性心房颤动患者心脏逆重构的影响. 安徽医科大学学报, 2015, 50: 223-226.
2014-09-03)
(编辑:梅 平)
国家自然科学基金(81471408)
400037 重庆市,中国人民解放军第三军医大学第二附属医院 心血管外科
易茜 硕士研究生 主要从事心脏疾病外科监护治疗和心房颤动结构重构的分子机制方面的研究 Email: danlanruozi@163.com 通讯作者:马瑞彦Email:maruiyan2008@hotmail.com
R541.4
A
1000-3614(2015)08-0813-04
10.3969/j.issn.1000-3614.2015.08.024
猜你喜欢
红蜻蜓·中年级(2024年5期)2024-06-14 09:11:51
保健医苑(2023年2期)2023-03-15 09:02:50
传染病信息(2022年3期)2022-07-15 08:24:28
趣味(作文与阅读)(2021年12期)2021-04-19 12:16:54
肝博士(2021年1期)2021-03-29 02:32:16
戏剧之家(2018年35期)2018-02-22 12:32:40
大众健康(2017年8期)2017-08-23 21:18:22
老友(2017年7期)2017-08-22 02:36:30
作文评点报·高中版(2017年9期)2017-03-27 13:20:15
医学研究杂志(2015年6期)2015-07-01 17:40:08
