
四核簇状和一维链状丁基锡α-萘乙酸酯的合成、结构及抗癌活性
2015-12-15 07:18冯泳兰庾江喜邝代治谭宇星张复兴蒋伍玖朱小明郑建华
无机化学学报 2015年4期
冯泳兰 庾江喜 邝代治*, 谭宇星,2 张复兴 蒋伍玖 朱小明 郑建华
(1衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008) (2湖南师范大学化学化工学院,长沙410081)
四核簇状和一维链状丁基锡α-萘乙酸酯的合成、结构及抗癌活性
冯泳兰1庾江喜1邝代治*,1谭宇星1,2张复兴1蒋伍玖1朱小明1郑建华1
(1衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008) (2湖南师范大学化学化工学院,长沙410081)
在相同条件下,α-萘乙酸分别与二丁基氧化锡、氧化双(三丁基锡)反应,合成了四核簇状二丁基锡α-萘乙酸酯{[n-Bu2Sn (O2CCH2C10H7)]2O}2(1)和一维链状三丁基锡α-萘乙酸酯[n-Bu3Sn(O2CCH2C10H7)]n(2)。经IR、1H和13C NMR、元素分析和X-射线单晶衍射表征结构。1具有核心结构为氧原子桥联锡构筑的Sn2O2四元环,且四元环二聚形成Sn4O4簇状,中间Sn2O2环的中心为整个分子的对称中心。1中,两相邻配合物分子间通过C-H…π作用、形成一维带状结构。配合物2中,锡原子通过羧基双齿桥联、组成一维链状配位聚合物。测试表明:1和2分别在250、175℃以下可以稳定存在;对人癌细胞Colo205、HepG2、MCF-7、Hela、NCI-H460增殖均有较强的抑制作用,且2的活性高于1。
有机锡;溶剂热合成;晶体结构;抗癌活性
有机锡因具有杀虫、抑菌、抗肿瘤等生物活性而引起人们的研究兴趣[1-3]。利用烃基锡卤化物的控制性水解或通过适当的烃基锡前体与含氧(氮、硫)配体反应,可组装成不同类型的新型有机锡化合物,一些由Sn-O键构筑的有机锡簇合物和大环状有机锡化合物不断被合成[4-7]。研究表明,反应物的结构和反应条件对形成的有机锡配合物的结构及其性能有着重要影响[8-13]。因此,近年来人们对有机锡配合物的合成方法进行了许多探索,从传统方法[14]到固相合成法[15]、溶剂热法[16]、微波辐射合成[17]
等等。溶剂热法和微波辐射合成技术用于有机锡的合成,虽然伴随着发生脱烃基反应的可能[18-19],但这
2种方法具有节能环保和易于控制组装成配合物等优点,自有相关文献报道以来,就受到了有机锡研究者们的青睐,尤其成为定向合成有机锡氧簇合物的重要手段[20-21]。我们利用溶剂热法,将α-萘乙酸分别与二丁基氧化锡、氧化双(三丁基锡)在相同的条件下反应,合成了2个新的有机锡羧酸酯{[n-Bu2Sn (O2CCH2C10H7)]2O}2(1)和[n-Bu3Sn(O2CCH2C10H7)]n(2),
对其结构进行了表征,并研究了它们的热稳定性及抗癌活性。
1 实验部分
1.1 仪器和试剂
熔点用X4双目体视显微熔点测定仪(北京泰克仪器有限公司)测定,温度计未经校正。C、H含量用PE-2400元素分析仪(美国PE公司)测定。红外光谱用IR Prestige-21红外光谱仪(日本Shimadzu公司,4 000~400 cm-1)测定。核磁共振氢谱、碳谱分别用Bruker Avance 400和Bruker Avance 500核磁共振仪(瑞士Bruker公司,TMS为内标)测定。热重分析于TG 209 F3热重分析仪(德国Netzsch公司)上进行。
二丁基氧化锡(XSn>47%)购自南通艾德旺化工有限公司,氧化双(三丁基锡)(96%)购自上海晶纯生化科技股份有限公司,α-萘乙酸(化学纯)购自上海来泽精细化学品厂,卡铂(99%)、氘代氯仿(XD≥99.8%)购自百灵威科技有限公司,二甲基亚砜(DMSO)购自天津市科密欧化学试剂厂,甲醇购自中国·天津市巴斯夫化工有限公司,其余试剂为分析纯。人结肠癌细胞(Colo205)、人肝癌细胞(HepG2)、人乳腺癌细胞(MCF-7)、人宫颈癌细胞(Hela)、人肺癌细胞(NCI-H460)细胞株取自美国模式培养物集存库(ATCC),含10%胎牛血清的RPMI-1640培养基购自美国Gibico公司,胰蛋白酶(Trypsin)购自甘肃金盛生化制药有限公司。
1.2 配合物的合成
合成反应如图1所示。将1 mmol(0.186 g)α-萘乙酸,1 mmol(0.249 g)n-Bu2SnO或0.5 mmol(0.298 g)(n-Bu3Sn)2O加入到10 mL甲醇中,搅拌混合均匀,然后转移至25 mL带聚四氟乙烯内衬的不锈钢反应釜中,于120℃下反应3 d,再按5℃·h-1的速率程序降至室温,得配合物1或2。
配合物1:无色透明晶体0.321 g,收率75.5%。m.p.100~102℃。元素分析(C80H104O10Sn4),理论值(%):C 56.50,H 6.16;实测值(%):C 56.48,H 6.17。IR (KBr,cm-1):3 042(w),2 955(s),2 924(s),2 859(m),1 626(s),1 576(vs),1 510(w),1 460(w),1 422(w),1 393 (s),1 339(m),1 325(m),1 288(w),1 258(w),1 234(w),1 163(w),1 078(w),1 018(w),964(w),935(w),878(w),779(s),712(w),685(w),638(s),540(m),474(m),419(w)。1H NMR(CDCl3,500MHz),δ 7.31~8.03(m,28H,Ar-H),4.11(s,8H,ArCH2COO),0.74~1.63(m,72H,Bu-H)。13C NMR(CDCl3,125 MHz),δ 13.56(Me-C),26.62 (SnCH2-),27.05(SnCH2CH2C H2-),27.41(SnCH2C H2-),41.91(Ar C H2COO),124.21,125.54,125.65,125.80,125.99,127.58,127.88,128.62,132.40,133.89(Ar-C),172.42,177.38(-COO)。
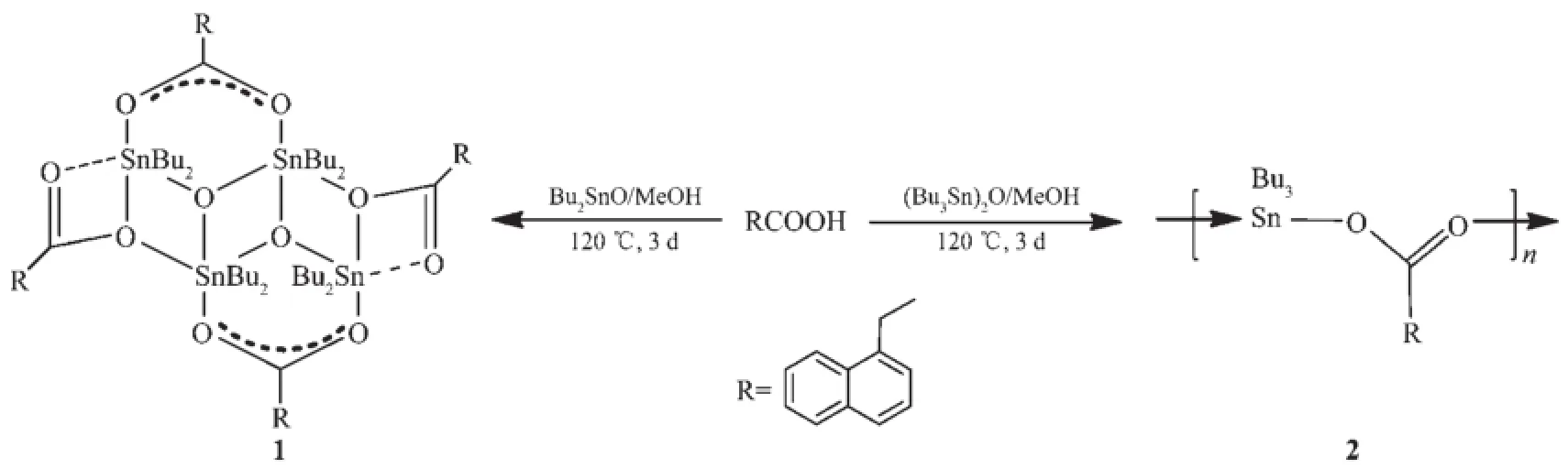
图1 配合物的合成反应Fig.1 Preparation of the comp lexes
配合物2:无色透明晶体0.402 g,收率84.6%。m.p.72~73℃。元素分析(C24H36O2Sn),理论值(%):C 60.65,H 7.64;实测值(%):C 60.63,H 7.65。IR(KBr,cm-1):3 051(w),2 955(s),2 920(m),2 853(m),1 583 (s),1 560(vs),1 510(w),1 464(w),1 420(w),1 389(m),1 290(w),1 258(w),1 180(w),1 157(w),1 076(w),1 018, (w),964(w),939(w),878(w),777(s),745(w),696(w),675 (w),631(w),611(w),583(w),544(w),424(w)。1H NMR (CDCl3,500MHz),δ 7.43~8.07(m,7H,Ar-H),4.08(s,2H,ArCH2COO),1.22~1.67(m,18H,-CH2CH2CH2-),0.92 (t,J=7.2 Hz,9H,-CH3)。13C NMR(CDCl3,100 MHz),δ 13.57(Me-C),16.52(SnCH2-,1J(119Sn-13C)=354 Hz,1J(117Sn -13C)=338 Hz),26.94(SnCH2CH2C H2-,3J(119Sn-13C)=65 Hz,3J(117Sn-13C)=61 Hz),27.74(SnCH2C H2-,2J(119Sn-13C) =21 Hz),40.13(Ar C H2COO),124.28,125.50,125.90,127.51,127.74,128.57,132.37,133.90,140.37,149.69 (Ar-C),174.09(-COO)。
1.3 晶体结构测定
选取尺寸为0.24 mm×0.18 mm×0.17 mm(1)和0.26 mm×0.17 mm×0.12 mm(2)的晶体,在Bruker SMART APEX II CCD单晶衍射仪上,采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),于296(2)K,以φ~ω扫描方式收集衍射数据。分别在2.92°~27.54° (1)和2.57°~27.46°(2)范围内收集衍射点。全部数据经Lp因子和多重扫描吸收校正。晶体结构由直接法解出,全部非氢原子坐标在差值Fourier合成中陆续确定,氢原子由理论加氢法给出在晶胞中的位置坐标。对氢原子和非氢原子分别采用各向同性和各向异性热参数进行全矩阵最小二乘法修正。对1中无序的羧基O1原子通过设置自由变量进行精修处理;对2中无序的3个丁基,通过限制相邻的C-C键长和间隔一个C的2个C原子间的距离,使其在合理范围内,并强制同1个丁基上无序的各C原子具有相似的原子位移因子进行精修。全部结构分析计算工作采用SHELXTL程序[22-23]完成。配合物的晶体学数据列于表1。
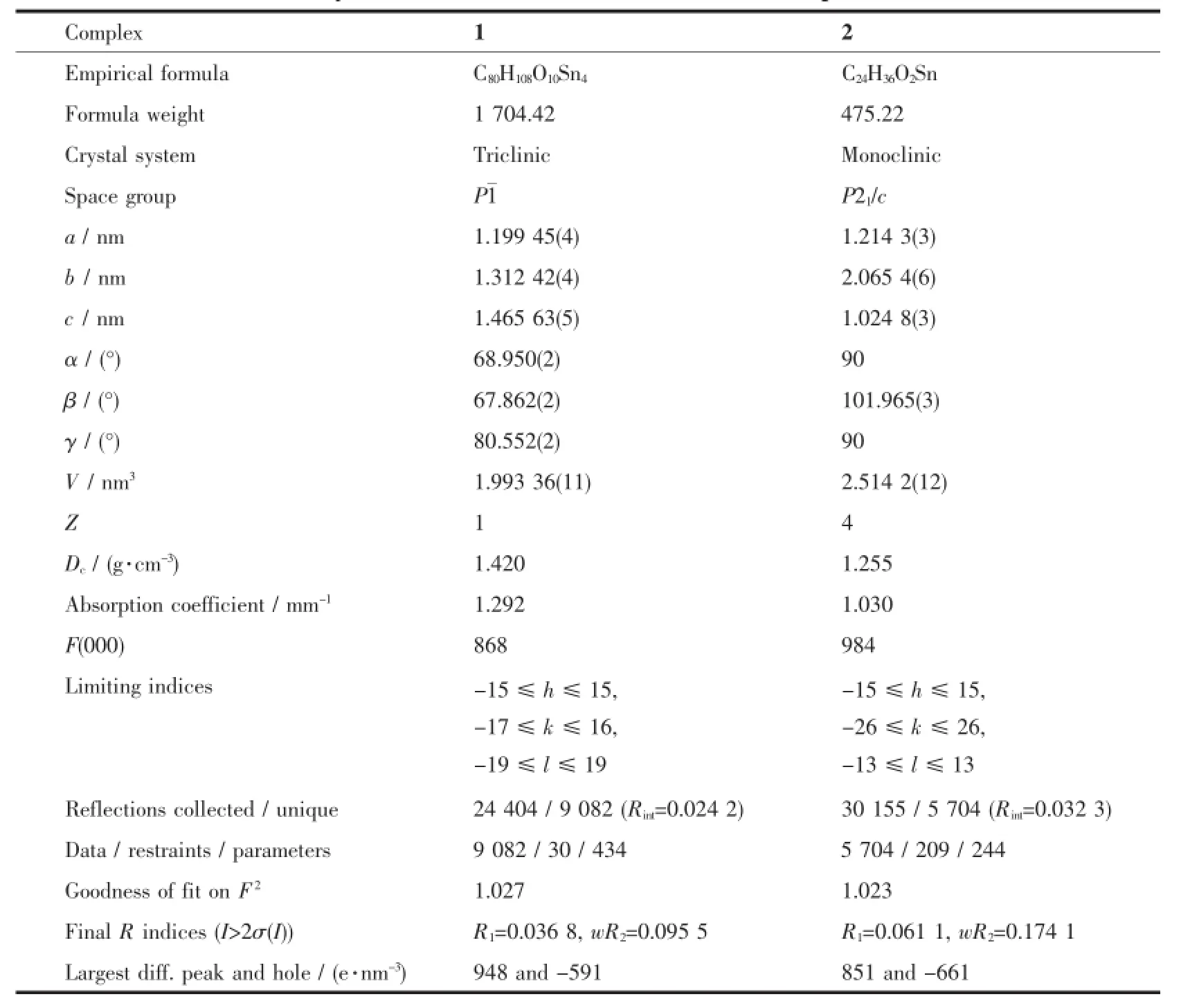
表1 配合物1和2的晶体学数据Table1 Crystal data and structure refinements of complexes 1 and 2
CCDC:1026774,1;1026775,2。
2 结果与讨论
2.1 谱学表征
配合物的红外光谱中,1(在2 955、2 924、2 859 cm-1)和2(在2 955、2 920、2 853 cm-1)均出现丁基的特征吸收,表明丁基存在。α-萘乙酸的羧羟基缔合吸收峰(2 500~3 300 cm-1)在配合物中消失,表明羧基去质子化与锡发生配位;α-萘乙酸在1 694、1 221 cm-1处的羧基不对称与对称伸缩振动,在配合物的IR中分别向低频和高频区迁移。1的羧基不对称伸缩振动出现在1 626、1 576 cm-1,对称伸缩振动出现在1 393、1 339 cm-1,其差值Δν分别为287和183 cm-1,说明1中存在以单氧形式和以双齿桥联形式与锡原子成键的2种类型的羧基[24-25],与晶体结构分析所得结果一致。2中羧基不对称与对称伸缩振动分别出现在1 560、1 389 cm-1,二者之差Δν为171 cm-1,表明羧基不对称与对称伸缩振动相向迁移更明显,羧基采用双齿配位。此外,1在638、474、419 cm-1出现Sn-O-Sn[26]、Sn-C、Sn-O伸缩振动吸收,2的Sn-C、Sn-O振动吸收出现在583、424 cm-1处[27]。
配合物的1H NMR谱中,各组峰的积分面积之比与预期的各组质子数基本吻合。1在7.31~8.03和2在7.43~8.07范围内的多重峰,归属为芳环质子吸收峰[1-3,5]。1在4.11和2在4.08处的单峰,归属为与芳环相连的亚甲基氢峰[28]。配合物的13C NMR谱中,芳环碳原子出现在124.21~133.89(1)和124.28~149.69(2)范围内。1在172.42和177.38处各出现1条谱线,表明1中羧基分2种类型,与红外光谱及单晶衍射实验结果相印证;2中羧基碳信号出现在174.09,与类似化合物的文献值[8,12,18,25]一致。
2.2 晶体结构
配合物1、2的分子结构分别见图2、3,主要键长和键角列于表2。从配合物的结构可知,相同的合成实验条件下,α-萘乙酸与Bu2SnO、(Bu3Sn)2O反应,其产物的结构有着明显的差异。
配合物1的核心结构为氧原子桥联锡构筑的Sn2O2四元环(由O3、Sn1、O4、Sn2组成),环中4个Sn-O键长不等且二面角∠O3-Sn1-O4-Sn2为2.149(86)°,该环为不规则平面四边形。2个这样的四元环利用Sn-O键稠合二聚形成Sn4O4簇状,中间Sn2O2环的中心就是整个分子的对称中心。4个羧酸酯基分为2类,一类以单氧形式桥联2个Sn原子,另一类则以它的2个氧原子以双齿桥联形式与Sn原子成键,在3个Sn2O2四元环两旁形成2个六元杂环。4个Sn原子除了与氧相互桥联外,每个Sn还连接2个丁基。由于锡周围的配基数及锡与各原子形成的键长、键角不等,锡有2类构型,Sn1、Sn1i为五配位变形三角双锥结构,Sn2、Sn2i为六配位变形八面体构型。另Sn1、Sn1i分别与O5、O5i之间存在着弱作用(Sn1与O5和Sn1i与O5i的距离为0.290 22(31)nm),因此Sn1、Sn1i也可被描述为六配位畸变八面体结构;而中间环的Sn2、Sn2i间还存在着Sn…Sn作用,Sn2与Sn2i的距离为0.331 92(3) nm。
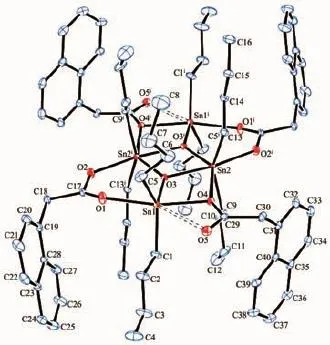
图2 配合物1的分子结构图(椭球率5%)Fig.2 Molecular structure of complex 1 with 5% probability ellipsoids

图3 配合物2的分子结构图(椭球率5%)Fig.3 Molecular structure of complex 2 with 5% probability ellipsoids
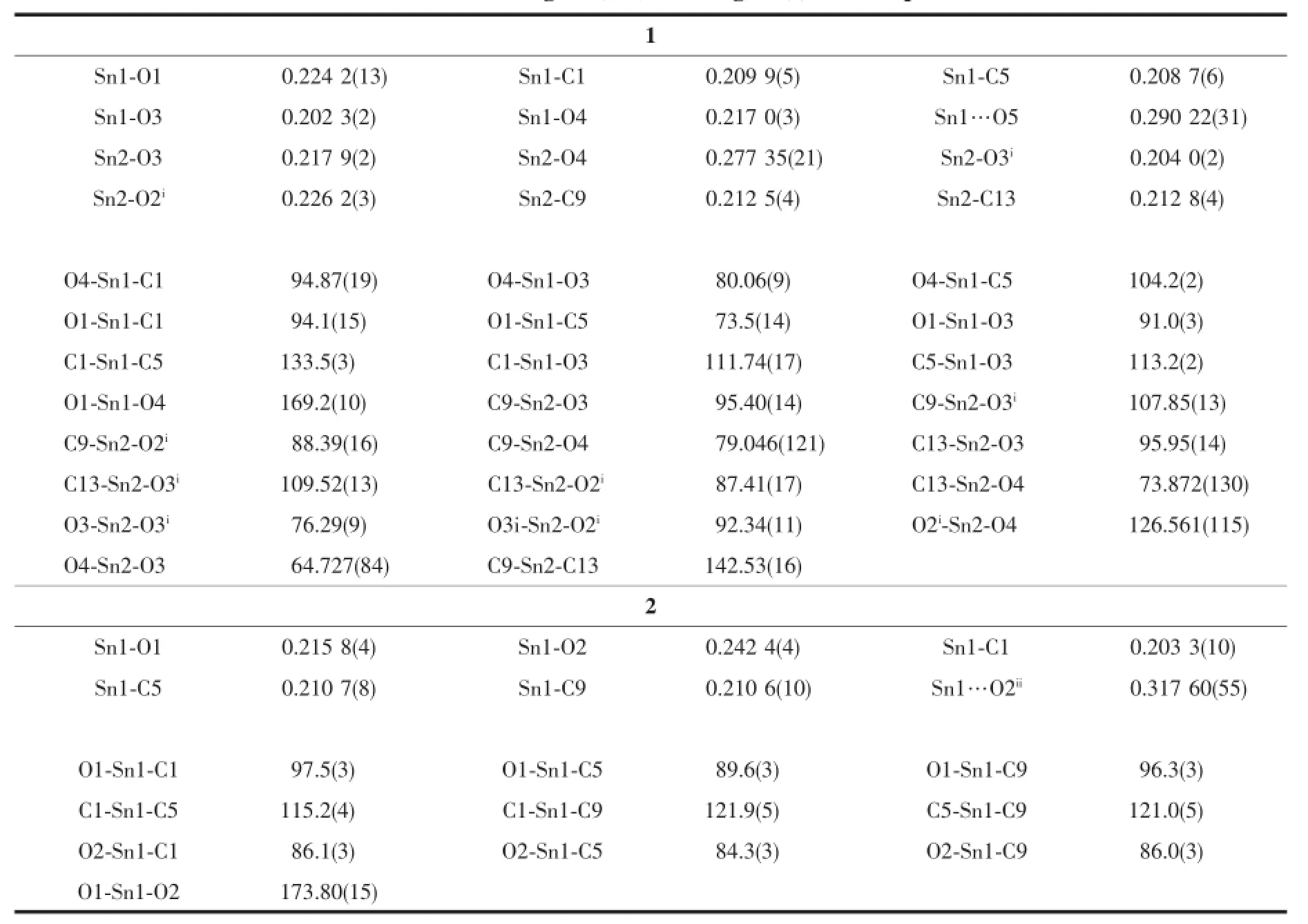
表2 配合物1和2的主要键长和键角Table2 Selected bond lengths(nm)and angles(°)for com p lexes 1 and 2
此外,在1的晶体中,2个相邻配合物分子间,通过丁基H8C与另一邻近分子的芳环之间发生C-H…π作用(dH8C…Cgi=0.296 71(1)nm,dC8…Cgi= 0.389 14(133)nm,∠C8-H8C…Cgi=161.971(928)°, Symmetry code:i-1+x,y,z)而成大环,且由这种弱作用把大环连接成一维带状结构(图4)。图4中,Cgi(Centroid:-0.092 17,0.609 93,0.794 30)代表1中苯环(由C23i~C28i原子组成)的质心。

图4 C-H…π作用构筑配合物1的一维带状结构Fig.4 1D ribbon structure of complex 1 by C-H…π interactions
配合物2中,可能由于锡原子上3个丁基的空间效应,未能形成四元环,Sn原子通过羧基双齿桥联,最终形成一维链状配位聚合物,如图5。在链中,中心锡原子与5个配位原子形成的键长、键角不同而组成五配位变形三角双锥,来自2个羧基的氧(O1、O2)占据三角双锥的轴向位置,轴向角∠O1-Sn1-O2为173.80(15)°,稍微偏离180°线性角,这可能与O2ii和Sn1间存在的弱作用(dSn1…O2ii= 0.317 60(55)nm)有关。
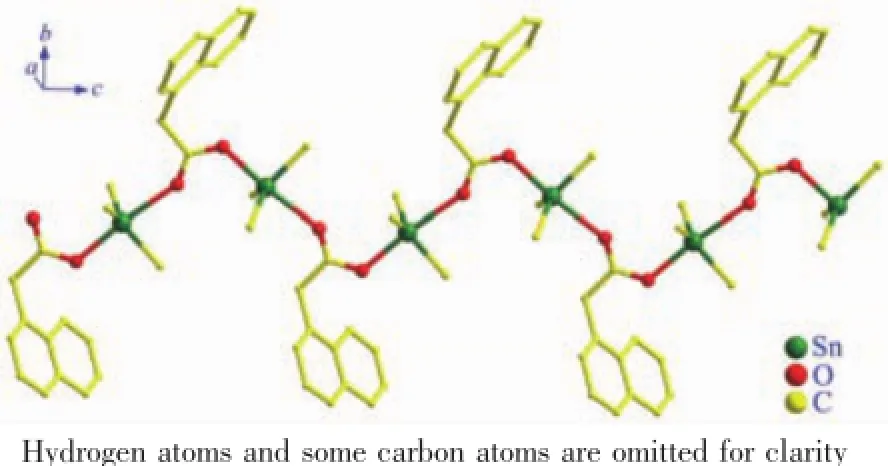
图5 配合物2一维链状结构Fig.5 1D chain structure of complex 2
2.3 热稳定性
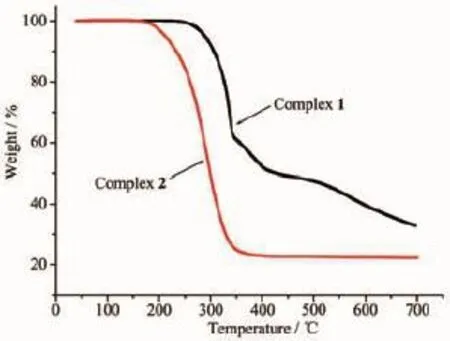
图6 配合物1和2的热重分析曲线Fig.6 Thermogravimetric analysis curves of complexes 1 and 2
为了解配合物的热稳定性,在空气氛下、以20℃·min-1的升温速率,于40~700℃范围内测得其热失重曲线,如图6所示。随着温度的上升,2个配合物均出现了较为明显的失重过程。1在250~375℃范围内,总重量损失了43.3%,对应于4个萘乙酸根的损失(计算值为43.6%);在375~695℃范围内又发生分解,失重23.7%,此时Sn-C键断裂、丁基脱离(计算值为26.9%);当温度高于695℃时,重量基本不再变化,此时重量共损失了67.0%。2在175~345℃范围内,总重量损失了74.3%。假定残渣对应的成分是SnO2,理论计算值分别为35.5%(1)和31.7% (2),计算值与实测值基本吻合。热重分析结果表明:尽管它们在较低温度熔化为液体,但1和2在较高温度仍具有良好的稳定性,分别在250、175℃以下可以稳定存在,它们在热稳定性上的明显差异与其结构的差异性有关。
2.4 抗癌活性
配合物的体外抗癌活性测试在中国科学院广州生物医药与健康研究院完成,与文献[3]同一方法,结果列于表3。1和2对人癌细胞Colo205、HepG2、MCF-7、Hela、NCI-H460增殖均有较好的抑制活性。1对人结肠癌细胞的抑制效果最好,其活性强于临床使用的卡铂;其次是对人肝癌细胞和人宫颈癌细胞;对人乳腺癌细胞和人肺癌细胞的抑制作用更次之,其活性弱于卡铂。配合物2对人结肠癌细胞的抑制作用最强,与1相当;对人肝癌细胞、人肺癌细胞、人乳腺癌细胞、人宫颈癌细胞的抑制活性依次减弱,但对这4种癌细胞的抑制效果仍远远高于配合物1和卡铂。配合物体外抗癌活性的测试结果表明,1和2均具有一定的药用价值。
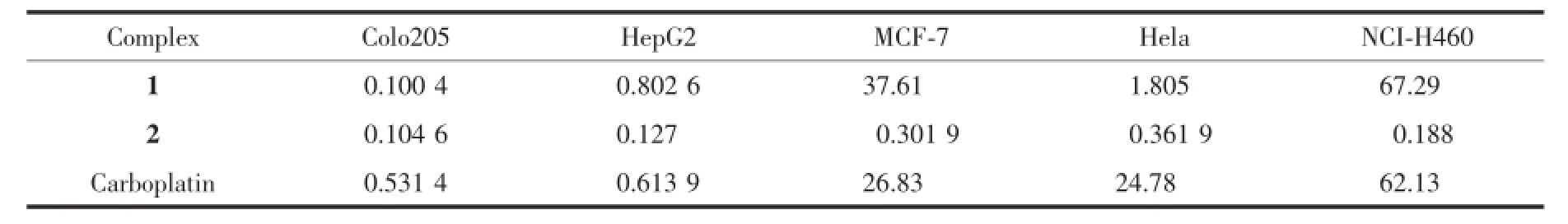
表3 配合物1、2和卡铂对肿瘤细胞的半抑制率Table3 IC50of com p lex 1,2 and carbop latin on tumor cells μmol·L-1
3 结论
α-萘乙酸与二丁基氧化锡、氧化双(三丁基锡)在相同的条件下反应,合成了2个新的有机锡羧酸酯1和2。1和2分别在250、175℃以下可以稳定存在;对人癌细胞Colo205、HepG2、MCF-7、Hela、NCI-H460增殖均有较强的抑制作用,且2的活性高于1。
[1]GONG Gen-Fei(龚根飞),SONG Ran(宋冉),ZOU Yin-Bo(邹引波),et al.Chinese J.Org.Chem.(有机化学),2013,33(4): 803-807
[2]Salam M A,Affan M A,Arafat M A,et al.Heteroatom Chem., 2013,24(1):43-52
[3]YU Jiang-Xi(庾江喜),FENG Yong-Lan(冯泳兰),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(6):1267-1272
[4]YU Jiang-Xi(庾江喜),FENG Yong-Lan(冯泳兰),PENG Yan (彭雁),et al.Chinese J.Inorg.Chem.(无机化学学报),2014, 30(5):1135-1142
[5]DENGYi-Fang(邓奕芳),CHENMan-Sheng(陈满生),ZHANG Chun-Hua(张春华),et al.Chinese J.Inorg.Chem.(无机化学学报),2009,25(12):2229-2232
[6]Chandrasekhar V,Mohapatra C,Butcher R J.Cryst.Growth Des.,2012,12(6):3285-3295
[7]Chandrasekhar V,Mohapatra C.Cryst.Growth Des.,2013,13 (11):4655-4658
[8]ZHANG Jun-Hong(张军红),ZHANG Ru-Fen(张茹芬),MA Chun-Lin(马春林),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(3):664-670
[9]Chandrasekhar V,Thirumoorthi R.Organometallics,2009,28 (7):2096-2106
[10]Prabusankar G,Murugavel R.Organometallics,2004,23(23): 5644-5647
[11]Chandrasekhar V,Metre R K,Biswas S.Organometallics, 2013,32(11):3419-3422
[12]Wang H,Li D,Hong M,et al.J.Organomet.Chem.,2013, 740:1-9
[13]Chandrasekhar V,Singh P.Organometallics,2009,28(17): 4974-4978
[14]TIAN Lai-Jin(田来进),WANG Xiao-Long(王晓龙),ZHENG Xiao-Liang(郑晓亮),et al.Chinese J.Inorg.Chem.(无机化学学报),2014,30(9):2087-2092
[15]Chandrasekhar V,Baskar V,Boomishankar R,et al. Organmetallics,2003,22(18):3710-3716
[16]Xie Y,Yang J,Ma J,et al.Cryst.Growth Des.,2009,9(9): 3881-3888
[17]Singh RV,Chaudhary P,Poonia K,et al.Spectrochim.Acta Part A,2008,70(3):587-594
[18]Wang Q,Ma C,He G,et al.Polyhedron,2013,49(1):177-182
[19]ZHANGFu-Xing(张复兴),WANG Jian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(3):537-543
[20]Xie Y,Yang J,Ma J,et al.Chem.Eur.J.,2008,14(13):4093 -4103
[21]Murugavel R,Gogoi N.J.Organomet.Chem.,2008,693(19): 3111-3116
[22]Sheldrick G M.SHELXS-97,Program for Crystal Structure Solution,University of Göttingen,Germany,1997.
[23]Sheldrick G M.SHELXL-97,Program for Crystal Structure Refinement,University of Göttingen,Germany,1997.
[24]YIN Han-Dong(尹汉东),XUE Sheng-Cai(薛绳才),WANG Qi-Bao(王其宝),et al.Chem.J.Chinese Universities(高等学校化学学报),2005,26(4):631-633
[25]Hong M,Yin H D,Zhang Y W,et al.J.Mol.Struct.,2013, 1036:244-251
[26]LU Yong-Quan(卢湧泉),DENG Zhen-Hua(邓振华).Practical Infrared Spectrum Analysis(实用红外光谱解析).Beijing: Electronics Industry Press,1989:242
[27]KE Yi-Kan(柯以侃),DONG Hui-Ru(董慧茹).Analysis Chemistry Handbook:Vol.2(分析化学手册:第三分册). Beijing:Chemical Industry Press,1998:932-935
[28]Muhammad N,Shah A,Rehman Z,et al.J.Organomet. Chem.,2009,694(21):3431-3437
Syntheses,Crystal Structures,and Anti-tumor Activity of Tetra-nuclear Cluster and 1D Chain Butyltin α-Naphthaleneacetic Carboxylates
FENG Yong-Lan1YU Jiang-Xi1KUANG Dai-Zhi*,1TAN Yu-Xing1,2ZHANG Fu-Xing1JIANG Wu-Jiu1ZHU Xiao-Ming1ZHENG Jian-Hua1
(1Key Laboratory of Functional Organometallic Materials of College of Hunan Province,Department of Chemistry and Material Science,Hengyang Normal University,Hengyang,Hunan 421008,China) (2College of Chemistry and Chem ical Engineering,Hunan Normal University,Changsha 410081,China)
The tetra-nuclear cluster dibutyltin α-naphthaleneacetic carboxylate{[n-Bu2Sn(O2CCH2C10H7)]2O}2(1) and the 1D chain tributyltin α-naphthaleneacetic carboxylate[n-Bu3Sn(O2CCH2C10H7)]n(2)were synthesized by the reactions of α-naphthaleneacetic acid with dibutyltin oxide and bis(tributyltin)oxide under the same conditions, respectively.Their structures were characterized by IR,1H and13C NMR,elemental analysis and X-ray crystal diffraction.The core structures of 1 are two Sn2O2four-membered rings which are constructed of Sn atoms with O bridges,and the two Sn2O2units are dimerized to a Sn4O4cluster,so the center of central Sn2O2ring coincides with the symmetry center of whole molecule.Due to the C-H…π interactions,the adjacent molecules of complex 1 is linked to generate 1D ribbon structure.For complex 2,two Sn atoms are bridged by two O atoms of a carboxylate ligand to form 1D infinite chain coordination polymer.Thermogravimetric analysis shows the complexes were stable up to 250 or 175℃,respectively.Moreover,the tests showed that both of them displayed strong in vitro anti-tumor activity against five human tumor cell lines,Colo205,HepG2,MCF-7,Hela and NCIH460,but the activity order is 2>1.CCDC:1026774,1;1026775,2.
organotin;solvothermal synthesis;crystal structure;anti-tumor activity
O614.43+2
A
1001-4861(2015)04-0710-07
10.11862/CJIC.2015.041
2014-10-27。收修改稿日期:2014-11-25。
湖南省自然科学基金(No.13JJ3112),湖南省科技计划(No.2014NK3086),湖南省高校创新平台开放基金(No.13K105,14K014,GN14K01),衡阳师范学院协同创新中心培育项目(No.12XT02)和青年骨干教师培养计划(2012),国家级大学生创业训练计划(No.201310546127)和
湖南省大学生研究性学习项目(No.CX1301)资助项目。
*通讯联系人。E-mail:hnkcq@qq.com;会员登记号:S06N8374M1012。
猜你喜欢
现代农村科技(2022年9期)2022-08-16
现代园艺(2020年3期)2020-03-05
山西化工(2019年3期)2019-08-01
广西林业科学(2016年2期)2016-03-20
应用化工(2014年1期)2014-08-16
应用化工(2014年3期)2014-08-16
应用化工(2014年10期)2014-08-16
应用化工(2014年7期)2014-08-09
应用技术学报(2014年1期)2014-02-28
