
HPLC法测定帕瑞昔布钠中有关物质的方法研究
2015-12-14 13:20聂忠莉郭兆元萧茂玲王晓玲
中国测试 2015年12期
聂忠莉,郭兆元,萧茂玲,王晓玲,张 勇,郭 瑞,叶 丁
(1.成都大学药学与生物工程学院,四川 成都 610106;2.成都大学四川抗生素工业研究所,四川 成都 610106;3.成都克莱蒙医药科技有限公司,四川 成都 610041)
HPLC法测定帕瑞昔布钠中有关物质的方法研究
聂忠莉1,郭兆元2,萧茂玲1,王晓玲3,张勇3,郭瑞3,叶丁3
(1.成都大学药学与生物工程学院,四川 成都 610106;2.成都大学四川抗生素工业研究所,四川 成都 610106;3.成都克莱蒙医药科技有限公司,四川 成都 610041)
建立以高效液相色谱法(HPLC)测定帕瑞昔布钠中有关物质的方法。色谱柱为C18柱(4.6mm×250mm×5μm),乙腈-0.01mol/L磷酸氢二钠溶液(用磷酸调节pH值至3.0)(47∶53,ν∶ν)为流动相,检测波长为215nm,柱温40℃。经过优化色谱条件后,帕瑞昔布钠峰与6个已知杂质峰均可达到基线分离,样品主峰及各杂质的色谱峰的灵敏度和分离度均满足测定要求。采用HPLC法测定帕瑞昔布钠中的有关物质,专属性强,方法灵敏度高,结果准确可靠。
HPLC法;帕瑞昔布钠;杂质;代地昔布
0 引 言
帕瑞昔布钠是一种治疗术后短期疼痛临床用药[1]。帕瑞昔布钠是伐地昔布的非活性前体药物,为水溶性药物。水解后,生成伐地昔布[4-(5-甲基-3-苯基异恶唑-4-基)苯磺酰胺]和丙酸,能高选择性地抑制COX-2[2],在发挥良好镇痛及抗炎作用[3]的同时,不影响胃肠道粘膜、血小板聚集及肾脏的功能。大量研究表明,帕瑞昔布钠用于骨科[4]、普外科[5]、妇科[6]等临床各科手术的术后镇痛效果良好[7],副作用较小,为需要经过非胃肠道给药的手术期患者带来了福音。
分析本品的合成工艺,并参考相关文献[8-9]及专利[10],本品中可能存在的有机杂质是:异噁唑(起始物料A)﹑中间体A、伐地昔布(中间体B);杂质A﹑B﹑C﹑D﹑E﹑F、G、H、I、J、K、L。本文采用高效液相色谱法对本品中可能存在的杂质E、H、J、K、L、伐地昔布(中间体B)和异噁唑(起始物料A)等有关物质进行检测和方法学验证研究,结果显示此方法专属性强,灵敏度高,适用于帕瑞昔布钠中有关物质的控制。其余的杂质不在本文研究之内。
1 仪器与试药
仪器:Series 1500高效液相色谱仪;UV759CRT紫外可见分光光度计;BP 210D电子分析天平;KH5200E型超声波清洗器(频率40 kHz,超声时长15 min);pHS-3C酸度计(上海精密科学仪器有限公司雷磁仪器厂)。
试药:帕瑞昔布钠(工作对照品批号:130801;测定样品批号:130903、130904、130905,成都克莱蒙医药科技有限公司,131101、131102、131103,成都通德药业有限公司);异噁唑、伐地昔布、杂质E、杂质J、杂质K、杂质H、杂质L对照品由成都克莱蒙医药科技有限公司研制;乙腈为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1溶液的配制
2.1.1供试品溶液
取帕瑞昔布钠样品适量,精密称定,用稀释剂乙腈-水(47∶53,ν∶ν)制成0.4mg/mL的供试品溶液。
2.1.2对照品溶液
精密量取供试品溶液适量,加稀释剂稀释至0.4μg/mL作为对照溶液。
取异噁唑、伐地昔布、杂质E、J、K、H、L对照品与帕瑞昔布钠,工作对照品各适量,精密称定,用稀释剂乙腈-水(47∶53,ν∶ν)溶解并稀释制成每1mL中含帕瑞昔布钠0.4mg、各杂质均为0.4μg的溶液,作为对照品溶液。
2.2测定波长的选择
取异噁唑、伐地昔布、杂质E、J、K、H、L对照品与帕瑞昔布钠对照品各适量,分别用乙腈-水(40∶60,ν∶ν)配置成0.02 mg/mL的溶液,照紫外-可见分光光度法[11]在200~400 nm范围内扫描[12],结果帕瑞昔布钠、中间体及各杂质均具有较大的紫外末端吸收,最终选择215 nm作为本品有关物质检测波长。

表1 帕瑞昔布钠有关物质分析方法色谱条件比较

图1 帕瑞昔布钠及杂质对照品定位色谱图
2.3色谱条件的确定
参照注射用帕瑞昔布钠进口注册标准[13],对帕瑞昔布钠有关物质进行分离考察,并对色谱条件进行不断优化,其结果见表1。
因此,本文选用色谱条件为C18柱(4.6 mm× 250mm×5μm),乙腈-0.01mol/L磷酸氢二钠溶液(用磷酸调节pH值至3.0)(47∶53,ν∶ν)为流动相,检测波长为215nm,柱温40℃。
2.4系统适用性试验及定位试验
取2.1.2项杂质对照品溶液10μL按2.3项下条件进样,记录色谱图,结果表明帕瑞昔布钠主峰与杂质对照品的杂质峰(包含杂质E、伐地昔布、J、K、H、L、异噁唑)以及溶剂空白之间的分离度均在2.4以上,能完全分离;其出峰结果见图1。

表2 有关物质强降解试验结果

图2 帕瑞昔布钠样品的专属性试验色谱图
2.5强降解试验
分别取帕瑞昔布钠样品适量,对其进行酸、碱、氧化、高温及紫外光照等强降解试验,按2.3项下条件进行测定,结果见表2和图2。
由上述实验结果可知:帕瑞昔布钠对热、酸、碱稳定,杂质基本无变化;对紫外灯光和氧化均不稳定,在不同的降解条件下,产生的杂质及杂质的变化量也不同;但最主要的变化量是氧化降解条件下,已知杂质中间体B(伐地昔布)达到了39.6%。在各降解条件下产生的杂质与主峰,以及杂质与杂质之间分离度均在1.5以上,能达到完全分离,主峰峰纯度均达到0.9999,杂质峰峰纯度基本均达0.99以上,峰降解平衡在95%~105%之间,结果表明该方法专属性良好,能检测出帕瑞昔布钠在储藏和运输过程的极端条件下产生的降解产物。
2.6检测限、定量限
取2.1.2帕瑞昔布钠对照品溶液适量,加乙腈-水(47∶53,ν∶ν)稀释至一定浓度,取此溶液适量按2.3项方法进样,记录色谱图,同时走空白基线,以仪器基线噪音3倍峰高作为检测限(S/N≥3),仪器基线噪音10倍峰高作为定量限(S/N≥10),得各杂质的定量限与检测限如表3所示。
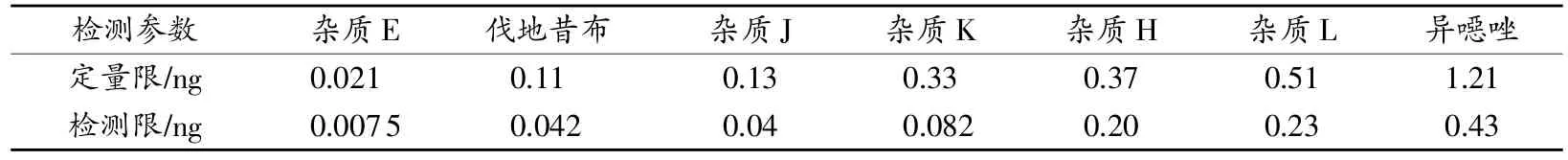
表3 帕瑞昔布钠有关物质检测限、定量限试验
2.7线性
精密称取帕瑞昔布钠及各杂质对照品适量,用乙腈-水(47∶53,ν∶ν)溶解并稀释至适宜浓度,精密量取各溶液10μL按2.3方法检测,记录色谱图。以浓度-峰面积做曲线,结果表明:在4.00~497.14μg/mL范围内,帕瑞昔布钠线性方程为Y=32.31x+16.04;r2=0.999 4;在0.10~5.0 μg/mL范围内,杂质E(Y=44.66x-1.60;r2=0.998 4)、伐地昔布(Y=42.397x+ 1.49;r2=0.9997)、杂质J(Y=43.42x+1.33;r2=0.9997)、杂质K(Y=34.60x+1.1;r2=0.9997)、杂质H(Y=21.19x+ 3.64;r2=0.9912)、杂质L(Y=35.20x+1.14;r2=0.9998)、异噁唑(Y=42.90x+1.09;r2=0.9998)色谱峰面积与其浓度均有良好的线性关系。
2.8重复性试验和中间精密度
6份帕瑞昔布钠样品,分别称取一定量并加乙腈-水(47∶53,ν∶ν)溶解稀释制成0.4mg/mL的供试品溶液,取此溶液适量按2.3项方法进样,记录色谱图。6份样品中,未知杂质按未加校正因子的自身对照法计算含量,已知杂质按峰面积外标法计算含量,结果显示6份样品均只检出伐地昔布和杂质K两个杂质,RSD分别为:4.77%、3.46%;其他杂质均未检出,重复性良好。
由不同人员在不同时间、不同仪器上对同一批号样品测定12次,各杂质的平均含量分别为:伐地昔布0.0128%、杂质K 0.0154%、总杂质0.0282%;RSD分别为:伐地昔布4.91%、杂质K 4.75%、总杂质4.34%,中间精密度良好。
2.9供试品溶液稳定性
取2.1.2项下对照品溶液,在室温下分别放置0,2,4,8,16,24h,按2.3方法检测,溶液在24h内都未产生任何新的杂质,如表4所示。

表4 帕瑞昔布钠有关物质稳定性试验(n=6)
2.10回收率试验
精密称定帕瑞昔布钠(130801)10mg(共10份)分别加入上述50%对照品溶液、100%对照品溶液、150%对照品溶液1.0mL各3份使样品溶解,分别作为50%、100%、150%的供试品溶液;剩余一份加入乙腈-水(47∶53,ν∶ν)使溶解,作为供试样品溶液。分别取上述对照品溶液、供试品溶液、供试样品溶液按2.3项方法进样,记录色谱图,回收率结果见表5。各杂质回收率良好。

表5 帕瑞昔布钠有关物质平均回收率
2.11耐用性
在检测波长、柱温、流动相流速、pH值、流动相比例发生微小变化的情况下,按2.3方法检测,样品中杂质个数和杂质量,以及各成分峰之间的分离度与原条件几乎无变化;但当色谱柱发生改变时,杂质H的出峰保留时间发生变化,且变化较大,但测定杂质含量变化不是很大,由此可证明在色谱柱不发生变化的情况下,本方法的耐用性良好;另从样品杂质检测情况看,各批样品中均未检测出杂质H,若本品中不存在杂质H的情况下,色谱柱的耐用性也可视为良好。
3 结果与讨论
3.1有关物质检测进样浓度的确定
根据化学药物研究技术指导原则对有关物质限度的要求,原料的杂质报告限度、鉴定限度、质控限度分别为0.05%、0.10%、0.15%(每日最大剂量<2g),本品每日的最大剂量为80 mg帕瑞昔布,因此为了保证0.05%的杂质能检出,根据本品的检测限试验结果,以进样10μL计算,有关物质检查的浓度至少应为0.43ng/10 μL/0.05%/103=0.086 mg/mL,参照帕瑞昔布钠进口注册质量标准有关物质检测的进样浓度及本品制剂规格,确定有关物质检测浓度为0.4mg/mL,进样量为10μL。
3.2色谱图记录时间的确定
由破坏试验结果可知,已知杂质峰及其破坏条件下产生的杂质峰均在主峰的3.5倍保留时间内,故确定供试品溶液色谱图记录时间为主峰保留时间的3.5倍。
4 结束语
通过上述实验结果表明:在优化的帕瑞昔布钠有关物质色谱条件下,破坏产生的杂质与主峰,以及杂质与杂质之间分离度均在1.5以上,能达到完全分离,主峰峰纯度均达到0.999 9,杂质峰峰纯度基本为0.99以上;各破坏样品的物料平衡均在95%~105%之间,说明该方法专属性良好,能检测出本品在储藏和运输过程的极端条件下产生的杂质。
[1]宋德玲.帕瑞昔布钠在术后镇痛中的应用进展[J].黑龙江医药,2013,26(3):481-482.
[2]催向丽,赵志刚,陈丽,等.新型注射用选择性COX-2抑制剂帕瑞昔布钠[J].中国新药杂志,2009,18(14):1283-1285.
[3]余雷霆,廖丽君,李顺,等.帕瑞昔布钠超前镇痛对老年腹腔镜胆囊切除术后应激和炎症反应的影响[J].浙江实用医学,2012,17(3):171-175.
[4]蒋宗明,陈忠华,郑羡河,等.帕瑞昔布钠超前镇痛对骨科患者术后血IL-1β、IL-6和IL-8水平的影响 [J].第三军医大学报,2010,32(13):1485-1486.
[5]黄福森,吴超然.帕瑞昔布钠联合头皮神经阻滞用于神经外科开颅术后镇痛的研究[J].重庆医科大学学报,2014,39(12):1809-1813.
[6]叶治,夏萍萍,王锷,等.不同超前镇痛时点应用帕瑞昔布钠对妇科手术后的镇痛效果[J].临床麻醉学杂志,2011,27(2):151-153.
[7]陈燕,董铁立.不同剂量的帕瑞昔布钠对全麻病人术中舒芬太尼用量的影响[D].河南:郑州大学,2012:13-17.
[8]王凯,徐泽彬,宋率华,等.帕瑞昔布的合成[J].中国医药工业杂志,2013(12):1207-1209.
[9]王凯,宋率华,金琪,等.帕瑞昔布钠合成路线图解[J].中国医药工业杂志,2013(8):836-838.
[10]Thakashinamoorthy C,Jesudoss M G,Hariharasubramania M,et al.A novel process for preparing valdecoxib:WO,2005085218[P].2005-09-15.
[11]国家药典委员会.中华人民共和国药典2010年版(二部)[S].北京:中国医药科技出版社,2010:29-31.
[12]Crane I M,Mulhern M G,Nema S.Stability of reconstituted parecoxib for injection with commonly used diluents[J].J Clin Pharm Ther,2003(28):363-369.
[13]JX20110120注射用帕瑞昔布钠进口注册标准[S].2011.
Research on HPLC method for related substances in parecoxib sodium
NIE Zhongli1,GUO Zhaoyuan2,XIAO Maoling1,WANG Xiaoling3,ZHANG Yong3,GUO Rui3,YE Ding3
(1.School of Pharmacy and Bioengineering,Chengdu University,Chengdu 610106,China;2.Sichuan Industrial Institute of Antibiotics,Chengdu University,Chengdu 610106,China;3.Chengdu Climb Pharmaceutical Technology Co.,Ltd.,Chengdu 610041,China)
To determine related substances in parecoxib sodium with high performance liquid chromatograph(HPLC).C18 column has been used as filler(4.6 mm×250 mm×5 μm)and acetonitrile-0.01 mol/L ammonium dihydrogen phosphate(adjusted to pH 3.0 with phosphoric acid)(47∶53,ν∶ν)as mobile phase.The detection wavelength was set at 215nm and the column temperature 40℃.After the optimization of chromatographic conditions,the parecoxib sodium peak and 6 known impurity peaks meet the requirement of baseline separation.The sensitivity and resolution of the main peak of the samples and the chromatographic peak of all the impurities are both acceptable.The HPLC method is specific and reliable.
HPLC method;parecoxib sodium;impurities;valdecoxib
A
1674-5124(2015)12-0054-05
10.11857/j.issn.1674-5124.2015.12.014
2015-04-01;
2015-06-06
聂忠莉(1966-),女,副主任药师,主要从事药物分析研究。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国药房(2022年10期)2022-05-30
色谱(2021年7期)2021-06-07
中华养生保健(2020年9期)2021-01-18
艺术品鉴(2020年6期)2020-12-06
首都食品与医药(2020年1期)2020-10-21
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
山东工业技术(2016年10期)2016-09-06
中学生数理化·中考版(2015年12期)2015-09-10
