
棉属四倍体AD1与二倍体A2、D5基因组的同源SSR分析
2015-12-02 02:11:55孙高飞何守朴潘兆娥杜雄明
遗传 2015年2期
孙高飞,何守朴,潘兆娥,杜雄明
棉属四倍体AD1与二倍体A2、D5基因组的同源SSR分析
孙高飞1,2,何守朴1,潘兆娥1,杜雄明1
1. 中国农业科学院棉花研究所,棉花生物学国家重点实验室,安阳455000;2. 安阳工学院计算机科学与信息工程学院,安阳455000
SSRs(Simple sequence repeats)是一类广泛存在于动植物基因组的DNA短串联重复序列,是重要的基因组分子标记。比较不同基因组同源SSR的差异,有利于了解相近物种间的进化过程。文章使用雷蒙德氏棉基因组(D5)、亚洲棉基因组(A2)全基因组序列和陆地棉(AD1)的限制性酶切基因组测序数据,进行全基因组SSR扫描,比较了A组和D组的SSR分布情况,通过识别3个基因组之间的同源SSR,比较它们之间同源SSR重复序列的差异。结果发现,A组和D组同源SSR的分布规律非常相似,但A组与AD组的同源SSR保守性比D组与AD组同源SSR的保守性强。与AD组同源SSR相比,A组中重复序列长度增长的SSR数量约为长度缩短的SSR数量的5倍,在D组中这一比值约为3倍。可以推测,四倍体AD组在与A组、D组的平行进化过程中,由于基因组融合,导致SSR的重复序列长度变化速率与二倍体A、D组有差异,同时这种差异可能导致了AD组SSR重复序列长度在进化过程中与二倍体相比有变短的趋势。文章首次对3个棉花基因组的同源SSR进行了系统地比较,发现了同源SSR在棉属四倍体基因组和二倍体基因组中的显著差异,为进一步揭示棉属基因组的进化规律提供了基础。
SSR;棉花基因组;同源SSR;进化
简单序列重复(Simple sequence repeats, SSRs)又称微卫星(Microsatellites),是一类由几个碱基组成的基序串联重复而成的DNA序列,基序长度一般为1~6 bp,总长一般大于或等于10 bp,广泛分布于动植物基因组,是重要的基因组分子标记。SSR具有多态性强、长度小、易于快速检测等特点,主要应用于动植物的分子标记开发、遗传图谱构建、基因定位等理论和应用研究[1,2]。
SSR在生物进化研究中也扮演着重要角色,在大规模基因组测序开始之前,对于SSR的研究是通过PCR等实验方法获得同源位点SSR,通过检测序列长度的差异来分析物种间的遗传关系,研究范围覆盖动物[3~6]和植物[7~10]。随着测序技术的发展,完整测序的基因组越来越多,对于SSR的研究也就更加的全面和深入,基于物种之间、群体之间的SSR的比较研究不断涌现[11~16]。
棉花SSR分子标记的开发在近年获得了快速进步[17~19],成为棉花分子生物学研究中应用最为成功的分子标记之一,广泛地应用于棉花种质资源的遗传多样性[20~22]、重要农艺性状的QTL定位[23~25]和全基因组关联分析等领域[26~28]。然而由于缺乏参考基因组,同时已开发的SSR标记来源相对单一(大多数来源于纤维EST库),因而对于棉花SSR在整个基因组上的分布和变化规律依然缺乏宏观的认识和研究。
通常认为,现在栽培上所用的异源四倍体陆地棉(AD1)的两个亚基因组供体种为二倍体亚洲棉(A2)和雷蒙德氏棉(D5)[29]。近年来随着这两个二倍体基因组草图[30,31]和部分陆地棉基因组测序原始序列的公布[32],人们对这3个基因组的结构了解地更加深入,但作为序列变异重要来源之一的SSR,特别是同源SSR在3个基因组之间的比较尚未见报道。本研究利用生物信息学方法,对亚洲棉、雷蒙德氏棉和陆地棉3个基因组同源SSR进行系统的比较和分析,重点探讨了3个基因组之间同源SSR的差异,以及产生这些差异的可能原因。
1 材料和方法
1.1 数据来源
雷蒙德氏棉基因组序列(以下简称D组)以及相关注释下载自NCBI (http://www.ncbi.nlm.nih.gov/ assembly/519268/),亚洲棉基因组(以下简称A组)序列以及相关注释信息下载自http://cgp.genomics. org.cn/。由于之前报道的陆地棉遗传图谱编号和两个二倍体全基因组测序编号存在差异,为了更直观分析SSR的同源性,本文首先根据相关文献对这些编号进行了整合[33](表1)。陆地棉基因组(以下简称AD组)测序数据来源于NCBI中http://www.ncbi.nlm.nih. gov/bioproject/168346。该基因组测序使用6种不同的陆地棉品种和两种不同的酶切方法,形成12个不同的测序序列样本,在本文中以下划线连接样本名称和内切酶名称做为陆地棉序列库的名称。
另外根据全基因组测序的染色体和基因注释信息,本文将基因组上的基因区域划分为外显子区(CDS)、内含子区(intron)、5ʹUTR区、3ʹUTR区、基因上游1000 bp(1 K)、基因下游1000 bp(1K)和非编码区(由于注释的原因,A组没有5ʹUTR区、3ʹUTR区)。使用perl语言编程,定位每个SSR在A组和D组上所在的基因区域,以便对各基因区域内包含的SSR数量进行比较。
<1),且各件产品是否为不合格品相互独立.
1.2 全基因组SSR扫描
基于Perl的MISA程序(http://pgrc.ipk-gatersleben.de/misa/)对A组、D组和AD组的基因组序列进行扫描,按照默认参数,识别最少为10次的单碱基重复和最少5次的2、3、4、5、6碱基重复为SSR。作为一个SSR认定的两个重复序列之间的碱基数不超过100 bp。
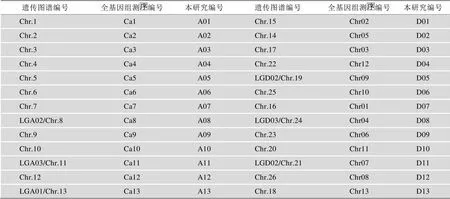
表1 本研究使用的棉花染色体编号与遗传图谱和基因组测序的染色体编号对比
1.3 同源SSR识别
将MISA扫描获得的数据分别建立SSR序列库。使用perl编写脚本分别提取A组、D组SSR基序两侧各100 bp碱基序列,形成长度超过210 bp的SSR识别序列。利用BLAST工具[34],将同源SSR识别序列映射到目标基因组进行匹配。由于BLAST的比对算法会产生不同长度的比对结果,在这些结果中,只有匹配长度达到一定的长度才能认为两个SSR识别序列是同源的。对于A组到D组、A组到AD组、D组到AD组的匹配记录,以匹配长度为观察参数,以10 bp为区间,分别计算匹配长度的分布(图1),可见,匹配长度在190 bp位置的分布数量迅速上升。考虑到侧翼序列有200 bp,取190 bp作为匹配长度,这时序列匹配度可达95%,因此选择190 bp作为认定SSR识别序列同源的阈值。如果A组的一个SSR (A_SSR)识别序列和D组的一段序列同源,则称这段序列是A_SSR识别序列在D组的同源匹配序列,如果该同源匹配序列中同时也存在一个SSR(D_SSR),则认为A_SSR和D_SSR为同源SSRs。从A组到D组识别同源SSRs,以及从A组到AD组、D组到AD组识别同源SSRs,均采用这一标准。
1.4 同源SSR的重复序列比对
通过MISA扫描获得的SSR重复类型有8种:c,c*,p1,p2,p3,p4,p5,p6。其中c和c*是组合型的SSR,即由两个或两个以上的重复序列组成。其中c*重复序列之间没有或只有一个其它碱基,而c类型的SSR重复序列之间包含若干个碱基。相对于组合型SSR,简单类型SSR是指由一个重复基序经过多次重复形成的重复序列,pn中的n是指基序的碱基数,例如p2值基序为2 bp的重复序列。
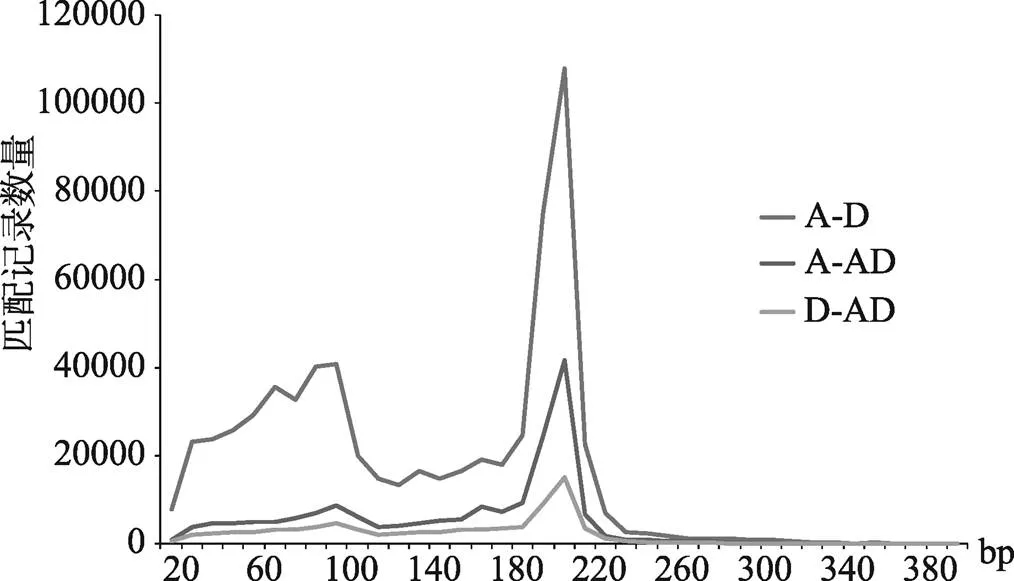
图1 3个基因组之间SSR识别序列Blast结果中不同匹配长度的数量分布
同源SSR中的重复序列总体上可以分为:重复类型不同和重复类型相同两种情况。本文将针对这两种不同的情况分别进行分析。
1.5 同源SSR类型差异统计
SSR变化的过程和机制目前还不清楚,因此,本文对于基因组之间同源SSR类型的差异情况只通过比较不同位置的SSR类型差异数量进行统计分析。
1.6 同源SSR类型相同的情况
对于类型相同的同源SSR,本文对匹配序列的A组和D组的SSR进行了分析,将两个SSR序列进行如下的分类:(1) 两个SSR的基序没有发生变化,只是重复基序的次数有变化;(2) 两个SSR的基序不同,但是这个不同是由于重复序列的起始位置的若干个碱基发生突变,导致重复基序产生了碱基的滑动,这种情况本文定义为基序移位,例如AGC和GCA,这种基序移位在比较和统计中认定为是基序相同;(3) 两个SSR的基序完全不同,例如AAT和GTC。
由于BLAST比对时,主要是以SSR两端的侧翼序列为识别序列,序列比对时已经确定了两个同源序列的匹配方向,因此SSR的方向应该和同源序列的方向是一致的。如果匹配序列的方向是反向的,则需将其中一个SSR基序的互补序列与另外一个SSR基序进行比较。例如基序为AAG和CTT的两个SSR,如果两翼序列匹配方向是反向的,则是完全相同的基序序列,如果两翼序列是正向匹配,则这两个SSR基序是完全不同的。在对同源SSR进行长度对比时,将基序不同的记录剔除,因为这样的记录中的两个SSR不是真正同源的SSR,很可能是在同源位点分别进化的SSR。
2 结果与分析
2.1 A基因组和D基因组SSR分布
以1Mb长度作为区间来研究SSR在A组和D组中的分布情况,同时根据两个基因组的注释,研究同区间的基因分布。结果发现,SSR和注释基因无论在位置还是数量上均高度一致,并且单位区间内SSR数量和基因数量高度相关(图2),相关系数分别为A (=0.913***) 和D (=0.939***)。
本文在A组中定位到326664个SSR (平均~4.69 kb一个),在D组中定位到191377个SSR (平均约~3.92 kb一个)。每条染色体中SSR的数量和其所在区域的长度高度相关(表2)。结果显示,外显子区的SSR数量明显低于其他区域,A组中的数量为2141个,而D组中SSR数量为2326个,但在与外显子区长度相当的内含子区域,却有将近10倍数量的SSR(A组为26 231个,D组为23 892个)。由于A组的UTR 区未注释,在进行比较时,将D组的5ʹ UTR数据合并至基因上游1 K,将3ʹ UTR数据合并至基因下游1 K。
由于SSR的数量和所在区域的长度有密切关系,为了更准确地了解SSR的分布规律,本文计算了A组、D组各基因区域的长度,SSR的数量和分布密度(表2)。由表2可知,CDS区SSR的密度最小,而在其他区域中,上游1 K的SSR密度最高,基因内含子和下游1 K的SSR密度也较高,这很可能是与SSR参与基因区域的转录调控有关[35]。
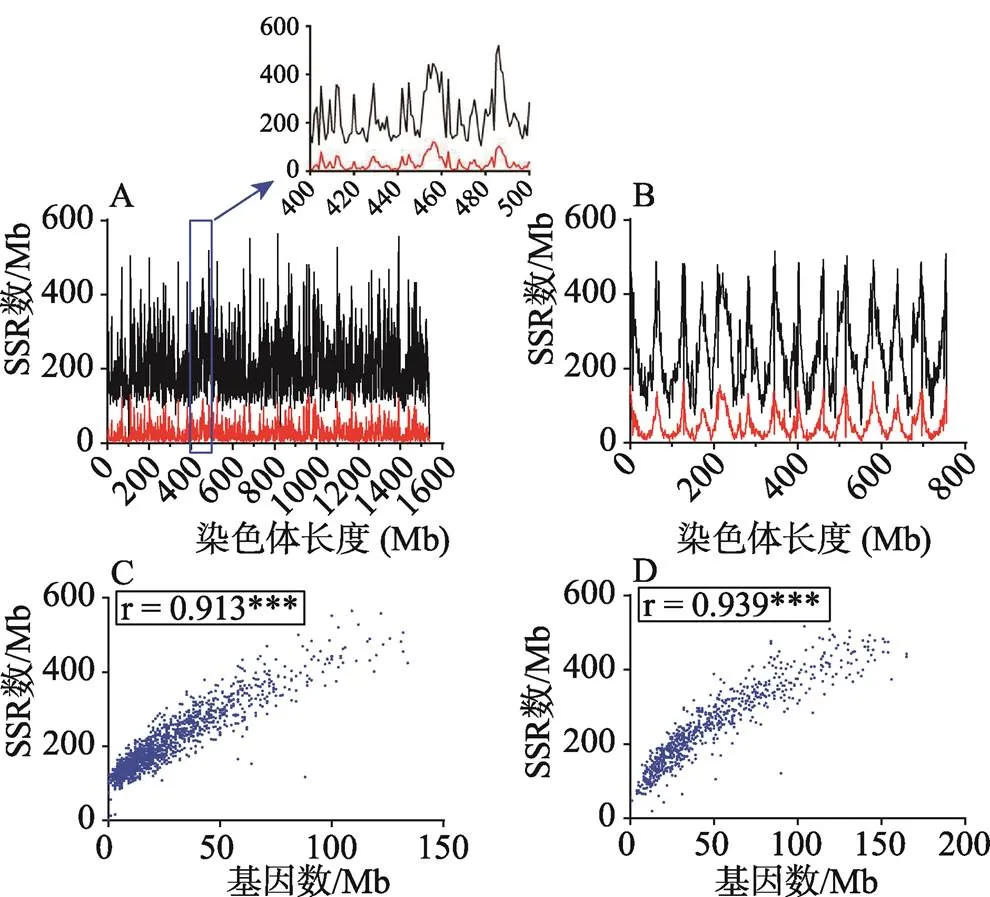
图2 A组、D组SSR分布图
A:A组SSR和基因在染色体上的分布;B:D组SSR和基因在染色体上的分布;C:A组单位区间内SSR数量和基因数量的相关性;D:D组单位区间内SSR数量和基因数量的相关性。
为了进一步了解不同基序类型SSR在基因组不同区域的分布特点,本文对A组、D组在各区域内不同基序类型SSR的数量所占比例进行了比较(图3)。A组、D组在基因上游1 K、基因下游1 K、基因内含子和非编码区的SSR类型分布非常一致,不同类型SSR所占比例从高到低依次是p1、p2、c、p3、p4、c*、p5和p6。CDS区的类型分布则和其他区域完全不同,p3类型占据了绝对的优势,这是因为在SSR的多态性类型中最常见的是重复序列的扩增和缩减,在CDS中除了p3和p6外,其他类型SSR的基序重复次数的变化,都可能导致基因阅读框的变化,从而改变基因翻译后的蛋白结构,因此会受到进化选择的限制。而且由于基序长度的限制,p6出现的概率明显小于p3,所以p3在CDS区的比例最大。另外一个略为突出的现象就是在非编码区,p2类型的比例比其他区域略高。
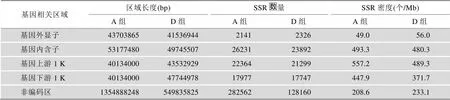
表2 A组、D组各基因区域SSR的数量与密度
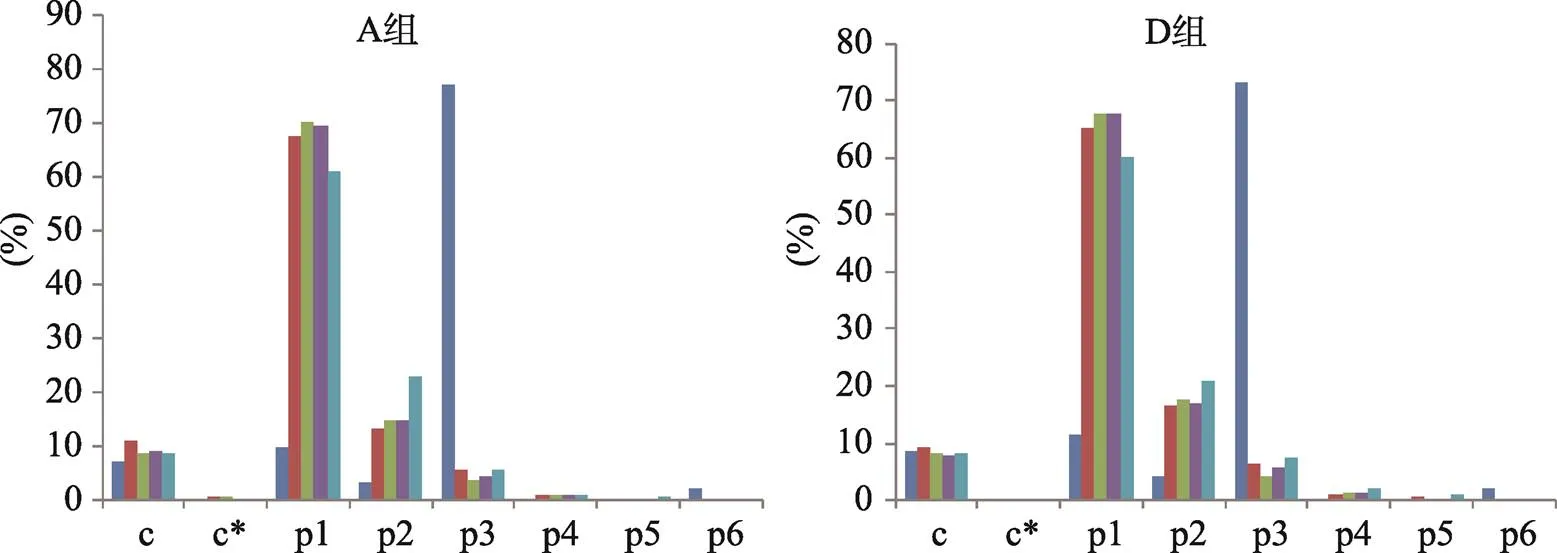
图3 不同基序类型SSR在A组、D组各基因区域的分布

2.2 A、D两个基因组中同源SSR的分析
2.2.1 A、D基因组同源SSR的染色体分布
通过同源SSR识别方法,在A组和D组中获得60037个同源SSR记录,每个记录都包含来自A组和D组的两个同源SSR,其中57275个同源记录都定位在组装的染色体上(部分定位在未组装的scaffold上)。根据同源记录在两个基因组染色体上的数量对应关系,本文构建了A组和D组SSR分布的关联图(图4)。
由于SSR在染色体上的分布相对均匀,SSR及侧翼序列可以看成基因组序列的部分抽样,同源SSR的对应关系和其在两个基因组上的分布能够部分反映两个基因组中染色体的同源性。从图4可以看出,在A组和D组的13个染色体中,大部分同源SSR分布于相应的同源染色体上,说明先前认定的同源染色体是可靠的。但是A01、A03、D01、D034个染色体之间有较大的同源SSR交换,尤其是A03和D01的同源SSR数量(1709)超过了A03和D03同源SSR的数量(1015),这说明染色体A03与D01之间SSR发生了大量的移位,从而使其同源性更高。这4条染色体的同源关系还有待进一步确认。
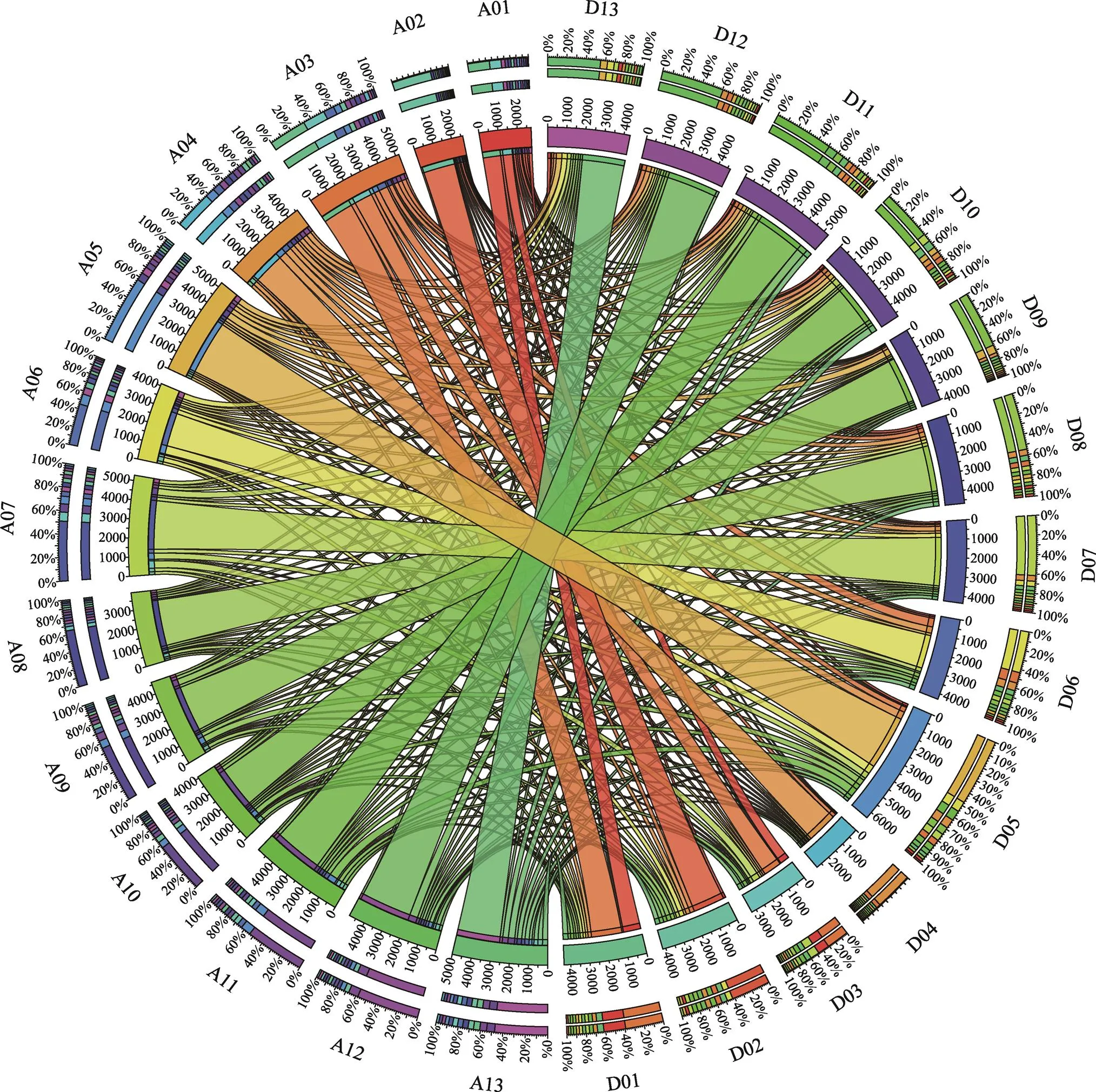
图4 A组和D组SSR分布关联图
2.2.2 A、D基因组同源SSR的比较
在A组和D组60037对同源SSR中,有10903对的重复类型不同。为了比较清晰地比较A组和D组之间的SSR的差异,本文将同源SSR记录分为两类:一是同源SSR重复类型不同的情况,一种是SSR重复类型相同的情况。在前者中重点比较SSR不同类型变化的数量,在后者中重点比较基序的长度比较。
在10903个同源SSR记录中,根据两个基因组不同的重复类型数量,比较重复类型差异比例(表3),可以看到组合型SSR的重复类型差异比例最大,简单型SSR基序碱基数量越大,重复类型差异的比例越大。A基因组中组合型SSR的重复类型差异比例高于D组,而D组的简单型SSR的重复类型差异比例均高于A组。
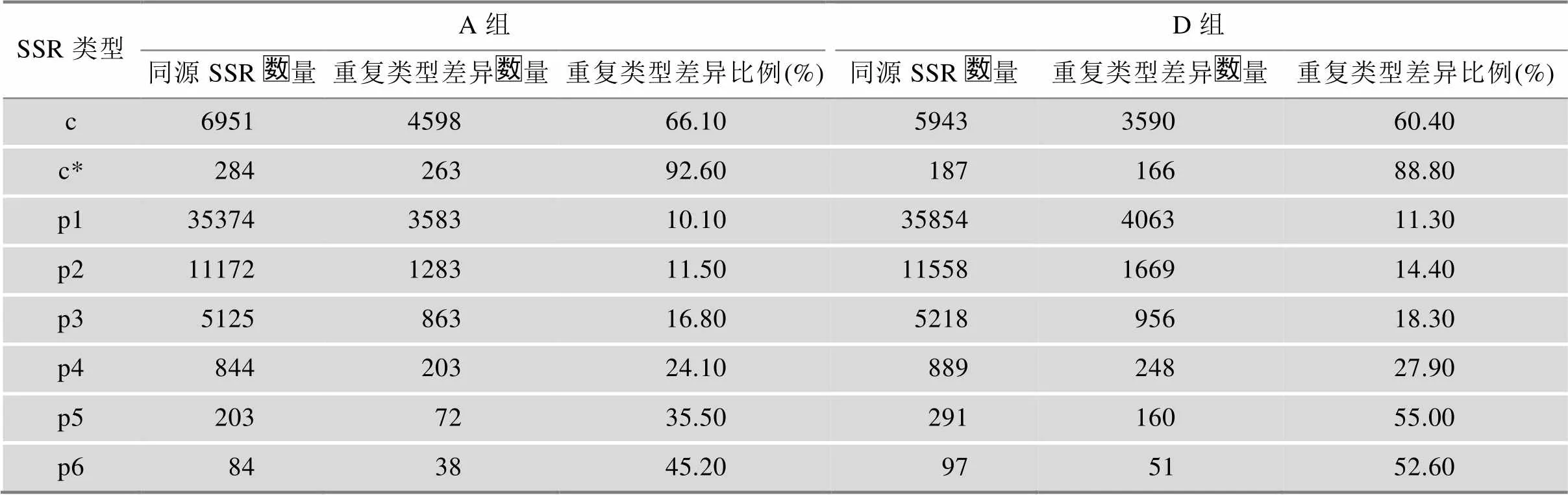
表3 A组和D组重复类型差异的同源SSR统计
本文将基因组不同位置的SSR重复类型差异进行了统计(表4),CDS区SSR重复类型差异的比例明显偏低,不足其他区域类型差异比例的一半。
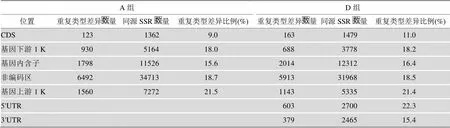
表4 A组和D组各基因区域重复类型差异的同源SSR分布
根据上述获得同源SSR的方法和基序比较方法,A组和D组SSR同源且类型相同的同源记录有49134条,去掉其中基序不同的2441条记录,对剩余的46693条基序相同的同源SSR记录进行重复序列长度的比较(表5)。比对A组和D组各基因区域同源SSR的重复序列长度,可以发现,在CDS区的SSR重复序列长度相等的比例是最大的,达到48.8%,在其他的位置,A组SSR重复序列长度大于D组SSR重复序列的数量,要比小于D组的比例多10%左右。
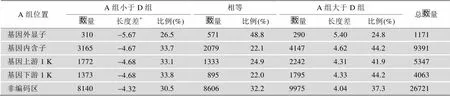
表5 A组与D组各基因区域的基序相同的同源SSR重复序列长度比较
注:*表中长度差为均值。
为了解基序相同的同源SSR的重复序列长度和重复类型是否存在关系,本研究比较了A组和D组各重复类型基序相同的同源SSR重复序列长度和重复类型(表6)。组合型的SSR长度相等的比例远低于其他类型,这说明组合型类型SSR在两个基因组间变化很大。在简单型SSR中,A组中p1类型SSR重复序列长度大于D组的数量远超过长度小于的数量(41.02%:28.52%),其他类型在A组SSR重复序列长度大于D组SSR和A组SSR重复序列长度小于D组,在比例上总体相当。不同重复类型SSR所表现的重复长度差异没有明显的规律。

表6 A组与D组各重复类型的基序相同的同源SSR长度比较
2.3 A组、D组和AD组SSR同源关系比较
2.3.1 总体比较
本研究使用A组SSR识别序列和D组SSR识别序列,分别对陆地棉(AD组)12个样本的测序序列进行了匹配定位,确定A组、D组和AD组的同源SSR。由于12个样本中的MCU_5_HpaII读段数量不足其他样本数量的1/3,导致与其他数据比较有较大差异,因此在进行统计时,剔除了样本MCU_5_HpaII的数据。
为了便于说明,本文将3个基因组之间的同源SSR关系绘制成图(图5),A组中和AD同源的SSR称为A-AD同源SSR;D组中和AD组同源的SSR,称为D-AD同源SSR。同理,AD组中和A组同源的SSR,称为AD-A同源SSR,AD组中和D组同源的SSR,称为AD-D同源SSR。
将AD组SSR分成3个部分:只和A组SSR同源的,称为AD-A特有同源SSR;只和D组SSR同源的,称为AD-D特有SSR;和A、D组均有同源SSR的称为共有同源AD组SSR。
AD-A同源SSR数量=AD-A特有同源SSR数量+共有同源AD组SSR数量;
AD-D同源SSR数量=AD-D特有同源SSR数量+共有同源AD组SSR数量;
依据以上的定义,A-AD同源SSR数量平均是D-AD同源SSR数量的2.76倍,而AD-A同源SSR的数量平均是AD-D组同源SSR数量的1.3倍,AD-A特有SSR的数量平均是AD-D特有SSR数量的1.61倍。
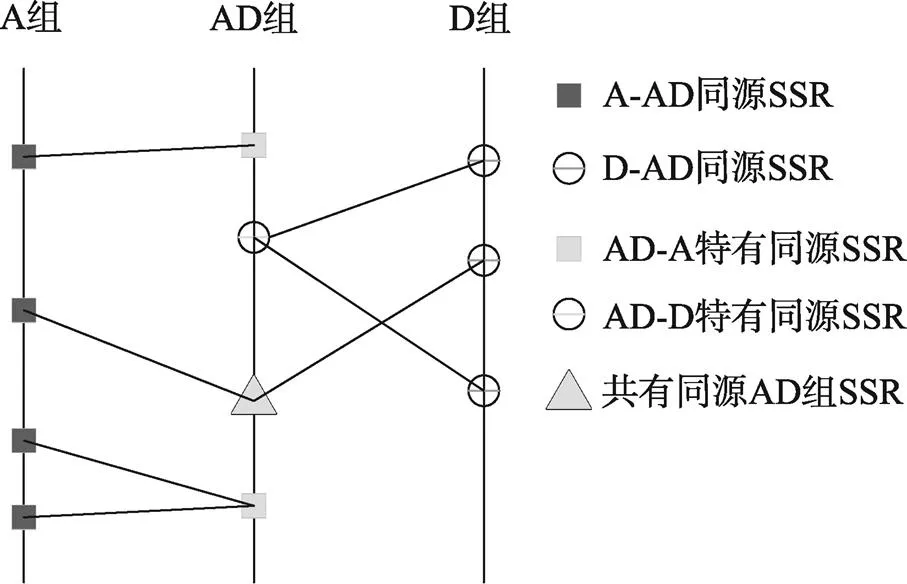
图5 3个基因组SSR同源关系图
2.3.2 3个基因组基序相同的同源SSR长度比较
分别对A组和AD组、D组和AD组的基序相同的同源SSR的重复序列长度进行了比较,计算了比较后A组、D组SSR重复序列长度小于、等于和大于AD组同源SSR记录的数量比例(表8),并分别计算了同源SSR之间的重复序列长度差的平均值。
为验证3个基因组之间基序相同的同源SSR长度差异是否显著,分别对3个基因组之间的SSR长度做了检验,其中A组和D组的同源SSR数量为60036对,检验值为3.97E-20,说明两个基因组SSR长度差异极为显著,A组和AD组、D组和AD组的SSR长度差异的显著性检验结果同样是极为显著(表8)。
A组和AD组同源SSR重复序列长度相等的数量高于D组和AD组同源SSR长度相等的数量比例,同时,A组和AD组同源SSR重复序列长度不同时的长度差小于D组和AD组同源SSR重复序列长度不同时的长度差。
本文对A、D组各基因区域的SSR和AD组同源SSR的重复序列长度比较结果进行分别统计(图6)。首先,无论A组和D组,重复序列长度大于AD组同源SSR的数量比例,远超过重复序列长度小于AD组同源SSR的数量比例;其次,在外显子区域,A、D组和AD组重复序列长度相等的同源SSR比例最高,说明SSR在外显子区域在3个棉种之间都非常保守;最后,A、D组重复序列长度小于AD组同源SSR的数量在各基因区域都比较接近。

表7 A组、D组和AD组同源SSR数量统计

表8 基序相同的同源SSR长度比较

图6 A组、D组和AD组各基因区域同源SSR重复序列长度对比
3 讨 论
本研究中3个基因组虽然同属于棉属,但是在基因组大小上有较大的差异,且AD基因组的测序方法与A组、D组的测序方法有所不同。要保证3个基因组间的SSR能够进行可信的比较,需要选取3个基因组中高度同源的SSR记录。
本研究使用SSR及两侧侧翼序列各100 bp作为识别序列到目标基因组进行匹配,并要求匹配长度大于等于190 bp,这一匹配要求是非常严格的(实验使用的PCR引物长度一般为20~30 bp),满足匹配条件的两个识别序列具有高度的同源性。同时对于匹配到的同源序列,要求其必须同样具有SSR重复序列,这进一步保证了匹配序列为高度同源的SSR序列。由于比较只在同源SSR间进行,因此能够避免基因组大小、染色体倍数和测序手段差异对研究结果的影响,最大限度保证了研究结果的可靠性。
在对同源SSR记录进行重复序列长度比较之前,先对SSR基序进行了比对,剔除了基序不同的同源SSR记录,因此,最后进行SSR重复序列长度比较的同源SSR记录,其最主要差异就是重复序列长度的差异,这部分同源记录中的SSR重复序列长度的变化保留了3个基因组在进化过程中留下的SSR重复序列长度变化的痕迹。
3.1 3个基因组CDS区的SSR非常保守
比较A组和D组SSR发现,无论在不同基因区域的SSR密度,还是不同重复类型SSR在基因区域所占的比例都非常接近。A组和D组CDS区重复类型有差异的SSR比例不到其他基因区域比例的一半(表4),在基序相同的同源SSR中,CDS区域长度相等的比例是其他区域的两倍多(表5),这些数据均说明A组和D组在CDS区域的同源SSR比其他区域保守得多。
A组和AD组比较,CDS区域长度相等的SSR所占比例为63.6%,D组和AD组比较,同源长度相等SSR所占比例为57.8%,约是其他区域的两倍(图6)。这也说明,3个基因组CDS区域SSR的保守型都要远高于其他区域。
同时,各基因组CDS区域的SSR数量明显小于其他区域。以上现象可能是因为SSR的高度可变性容易导致外显子区阅读框的改变,从而影响基因的功能,由于进化的选择,大量CDS区域的SSR被淘汰并趋于稳定,从而导致外显子区SSR的高度保守[36,37],这与已有研究的结果是相符的[8,38]。
3.2 就同源SSR数量和长度变化而言,A组在同源性上比D组更接近AD组
A组的SSR数量约是D组SSR数量的1.8倍(表2),A-AD同源SSR数量约是D-AD同源SSR数量的2.76倍(表7),这说明和D组相比,A组中SSR中有更高的比例与AD组同源的SSR。
同样,AD-A组同源SSR是AD-D同源SSR的1.3倍,而特有的AD-A同源SSR是特有AD-D同源SSR的1.6倍(表7),这说明AD组和A组同源的SSR数量明显高于和D组同源SSR数量。A组和D组的SSR的分布规律是非常相似的,因此,如果把SSR所在序列看成基因组的抽样,可以推测,A组和AD组的SSR同源性高于D组和AD组的SSR同源性。另外,在同源且类型相同的SSR记录中,A组和AD组同源SSR长度相同的比例(41.8%)要高于D组和AD组同源SSR长度相同的比例(37.5%)(表8),这说明A组和AD组的同源SSR保守性比D组和AD组的同源SSR保守性更高,这从另一个方面也印证了上述结论。
A组小于AD组同源SSR的平均长度差为-4.15,而D组小于AD组同源SSR的平均长度差为-4.81;A组大于AD组同源SSR的平均长度差为6.4,而D组大于AD组同源SSR的平均长度差为7.72(表8)。D组与AD组的同源SSR长度差均大于A组和AD组同源SSR的长度差,这也是A组与AD组的SSR同源性高于D组与AD组的SSR同源性的又一个佐证。
3.3 AD组和A、D组相同基序的同源SSR长度变化的数量差异
A组和D组来自共同的祖先,而AD组是A组和D组融合加倍而成,3个基因组是平行进化的[30]。根据同源SSR的长度比较,A组小于D组和A组大于D组的SSR数量比例基本是相等的(表6),但是,A组SSR长度大于AD组同源SSR长度的SSR数量,约是A组SSR长度小于AD组同源SSR长度的SSR数量的5倍,D组和AD组比较的结果约为3倍(表8)。产生这种情况,有两种可能:一种情况是A组和D组相对于AD组,大量SSR的长度增长了;另一种是AD组和A组、D组相比,大量SSR的长度缩短了。无论是哪一种情况,都可以认为,AD组的SSR长度变化速率与A、D组是不同的,而且,这种变化具有倾向性(多数SSR倾向于增长,或者缩短),因为只有这种倾向性,才有可能导致目前本文获得的3个基因组之间SSR长度差异情况。
关于人和黑猩猩[39]、羊和牛[3]的相近物种SSR长度差异现象很早就被提出,之后这种差异被解释为是由于测量偏差(Ascertainment bias)而导致的,同时也有观点认为测量偏差不能完全解释人和黑猩猩之间SSR的长度差异[40]。后来又有研究发现,人类除了两碱基重复的SSR的长度明显长于黑猩猩之外,其他重复类型的SSR没有发现明显的差异,而单碱基重复还发现了相反的趋势[4]。上述研究从不同的角度研究了相近物种之间的同源SSR长度差异,认为相近物种之间导致SSR长度差异的原因很可能是因为突变速率存在差异[11]。
由于本研究直接使用SSR侧翼序列匹配来获得同源SSR,侧翼两端序列长度取值达到200 bp,匹配长度不小于190 bp,因此获得的同源SSR序列的同源碱基比例超过95%。同时,本研究是使用统一的条件来获得3个基因组内所有的同源SSR,不属于抽样调查,因此本身可以排除测量偏差。在高度同源的SSR对比中,AD组的SSR长度与A组、D组相比,变长的SSR数量远低于变短的SSR数量。考虑到A、D组的SSR长度差异的数量比例比较接近,而且它们都是二倍体,而AD组是四倍体,这是和A、D组最为明显的差异。因此这种SSR长度变化在数量的差异,很有可能与AD组是四倍体而A、D基因组是二倍体有关。本文推测由于AD组是A、D基因组的融合,而这种融合过程导致了SSR长度变化速率的差异,而且这种差异导致了SSR长度变化的倾向性(大部分的SSR倾向于增长或缩短),进而形成了现有的AD组中大量的SSR长度小于A、D组同源SSR的现象。
在进化过程中,SSR的长度受复制滑动事件和点突变等多因素影响[41,42],因此A、D组中大量SSR相对于AD组同源SSR同步增长的概率要比AD组部分SSR的长度缩短的概率小,因此本文推测,四倍体的棉花AD组SSR相对于二倍体的A、D组同源SSR具有长度变短的倾向性。
[1] Ellegren H. Microsatellites: simple sequences with complex evolution., 2004, 5(6): 435–445.
[2] Morgante M, Olivieri AM. PCR-amplified microsatellites as markers in plant genetics., 1993, 3(1): 175–182. Ellegren H, Moore S, Robinson N, Byrne K, Ward W, Sheldon BC. Microsatellite evolution-a reciprocal study of repeat lengths at homologous loci in cattle and sheep., 1997, 14(8): 854–860.
[3] Webster MT, Smith NGC, Ellegren H. Microsatellite evolution inferred from human-chimpanzee genomic sequence alignments., 2002, 99(13): 8748– 8753.
[4] Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. High resolution of human evolutionary trees with polymorphic microsatellites., 1994, 368(6470): 455–457.
[5] 杨弘, 李大宇, 曹祥, 邹芝英, 肖炜, 祝璟琳. 微卫星标记分析罗非鱼群体的遗传潜力. 遗传, 2011, 33(7): 768–775.
[6] Peakall R, Gilmore S, Keys W, Morgante M, Rafalski A. Cross-species amplification of soybean () simple sequence repeats (SSRs) within the genus and other legume genera: implications for the transferability of SSRs in plants., 1998, 15(10): 1275–1287.
[7] Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes., 2002, 30(2): 194–200.
[8] Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S. Computational and experimental analysis of microsatellites in rice (L.): frequency, length variation, transposon associations, and genetic marker potential., 2001, 11(8): 1441–1452.
[9] 谢文刚, 张新全, 马啸, 彭燕, 黄琳凯. 鸭茅种质遗传变异及亲缘关系的SSR分析. 遗传, 2009, 31(6): 654–662.
[10] Vowles EJ, Amos W. Quantifying ascertainment bias and species-specific length differences in human and chimpanzee microsatellites using genome sequences., 2006, 23(3): 598–607.
[11] Kelkar YD, Tyekucheva S, Chiaromonte F, Makova KD. The genome-wide determinants of human and chimpanzee microsatellite evolution., 2008, 18(1): 30–38. Chistiakov DA, Hellemans B, Volckaert FAM. Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics., 2006, 255(1–4): 1–29.
[12] La Rota M, Kantety RV, Yu JK, Sorrells ME. Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley., 2005, 6(1): 23.
[13] Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, Myers RM. Worldwide human relationships inferred from genome-wide patterns of variation., 2008, 319(5866): 1100–1104.
[14] Sonah H, Deshmukh RK, Sharma A, Singh VP, Gupta DK, Gacche RN, Rana JC, Singh NK, Sharma TR. Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in., 2011, 6(6): e21298.
[15] Wang HT, Li XM, Gao WH, Jin X, Zhang XL, Lin ZX. Comparison and development of EST-SSRs from two 454 sequencing libraries of., 2014, 198(2): 277–288. Han ZG, Wang CB, Song XL, Guo WZ, Gou ZY, Li CH, Chen XY, Zhang TZ. Characteristics, development and mapping ofderived EST-SSRs in allotetraploid cotton., 2006, 112(3): 430–439.
[16] Wang CB, Guo WZ, Cai CP, Zhang TZ. Characterization, development and exploitation of EST-derived microsatellites inUlbrich., 2006, 51(5): 557–561.
[17] Lacape JM, Dessauw D, Rajab M, Noyer JL, Hau B. Microsatellite diversity in tetraploid: assembling a highly informative genotyping set of cotton SSRs., 2007, 19(1): 45–58.
[18] Alves MF, Barroso PA, Ciampi AY, Hoffmann LV, Azevedo VC, Cavalcante U. Diversity and genetic structure among subpopulations of(Malvaceae)., 2013, 12(1): 597–609.
[19] Liu DQ, Guo XP, Lin ZX, Nie YC, Zhang XL. Genetic diversity of Asian cotton (L.) in China evaluated by microsatellite analysis., 2006, 53(6): 1145–1152.
[20] Shen XL, Zhang TZ, Guo WZ, Zhu XF, Zhang XY. Mapping fiber and yield QTLs with main, epistatic, and QTL× environment interaction effects in recombinant inbred lines of upland cotton., 2006, 46(1): 61–66.
[21] Mei M, Syed NH, Gao W, Thaxton PM, Smith CW, Stelly DM, Chen ZJ. Genetic mapping and QTL analysis of fiber-related traits in cotton ()., 2004, 108(2): 280–291.
[22] Jiang CX, Wright RJ, Woo SS, DelMonte TA, Paterson AH. QTL analysis of leaf morphology in tetraploid(cotton)., 2000, 100(3–4): 409–418.
[23] Jia YX, Sun XW, Sun JL, Pan Z, Wang XW, He SP, Xiao SH, Shi WJ, Zhou ZL, Pang BY, Wang LR, Liu JG, Ma J, Du XM, Zhu J. Association mapping for epistasis and environmental interaction of yield traits in 323 cotton cultivars under 9 different environments., 2014, 9(5): e95882.
[24] Abdurakhmonov IY, Kohel RJ, Yu JZ, Pepper AE, Abdullaev AA, Kushanov FN, Salakhutdinov IB, Buriev ZT, Saha S, Scheffler BE, Jenkins JN, Abdukarimov A. Molecular diversity and association mapping of fiber quality traits in exoticL. germplasm., 2008, 92(6): 478–487.
[25] Kantartzi SK, Stewart JM. Association analysis of fibre traits inaccessions., 2008, 127(2): 173–179.
[26] Wendel JF, Brubaker C, Alvarez I, Cronn R, Stewart JM. Evolution and natural history of the cotton genus. In: Paterson AH, ed. Genetics and Genomics of Cotton. US: Springer, 2009: 3–22.
[27] Li FG, Fan GY, Wang KB, Sun FM, Yuan YL, Song GL, Li Q, Ma ZY, Lu CR, Zou CS, Chen WB, Liang XM, Shang HH, Liu WQ, Shi CC, Xiao GH, Gou CY, Ye WW, Xu X, Zhang XY, Wei HL, Li ZF, Zhang GY, Wang JY, Liu K, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu SS. Genome sequence of the cultivated cotton., 2014, 46(6): 567–572.
[28] Wang KB, Wang ZW, Li FG, Ye WW, Wang JY, Song GL, Yue Z, Cong L, Shang HH, Zhu SL, Zou CS, Li Q, Yuan YL, Lu CR, Wei HL, Gou CY, Zheng ZQ, Yin Y, Zhang XY, Liu K, Wang B, Song C, Shi N, Kohel RJ, Percy RG, Yu JZ, Zhu YX, Wang J, Yu SX. The draft genome of a diploid cotton., 2012, 44(10): 1098–1103.
[29] Rai KM, Singh SK, Bhardwaj A, Kumar V, Lakhwani D, Srivastava A, Jena SN, Yadav HK, Bag SK, Sawant SV. Large-scale resource development inL. by 454 sequencing of genic-enriched libraries from six diverse genotypes., 2013, 11(8): 953–963.
[30] Wang K, Song XL, Han ZG, Guo WZ, Yu JZ, Sun J, Pan JJ, Kohel RJ, Zhang TZ. Complete assignment of the chromosomes ofL. by translocation and fluorescence in situ hybridization mapping., 2006, 113(1): 73–80. McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools., 2004, 32(Web Server issue): W20–W25.
[31] Sawaya S, Bagshaw A, Buschiazzo E, Kumar P, Chowdhury S, Black MA, Gemmell N. Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements., 2013, 8(2): e54710.
[32] Tóth G, Gáspári Z, Jurka J. Microsatellites in different eukaryotic genomes: survey and analysis., 2000, 10(7): 967–981.
[33] Loire E, Higuet D, Netter P, Achaz G. Evolution of coding microsatellites in primate genomes., 2013, 5(2): 283–295.
[34] Li YC, Korol AB, Fahima T, Nevo E. Microsatellites within genes: structure, function, and evolution., 2004, 21(6): 991–1007.
[35] Garza JC, Slatkin M, Freimer NB. Microsatellite allele frequencies in humans and chimpanzees, with implications for constraints on allele size., 1995, 12(4): 594–603.
[36] Cooper G, Rubinsztein DC, Amos W. Ascertainment bias cannot entirely account for human microsatellites being longer than their chimpanzee homologues., 1998, 7(9): 1425–1429.
[37] Kruglyak S, Durrett RT, Schug MD, Aquadro CF. Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations., 1998, 95(18): 10774–10778.
[38] Santibáñez-Koref MF, Gangeswaran R, Hancock JM. A relationship between lengths of microsatellites and nearby substitution rates in mammalian genomes., 2001, 18(11): 2119–2123.
(责任编委: 刘宝)
Homologous simple sequence repeats (SSRs) analysis in tetraploid (AD1) and diploid (A2, D5) genomes of
Gaofei Sun1,2, Shoupu He1, Zhaoe Pan, Xiongming Du1
Simple sequence repeats (SSRs)are a class of repetitive DNA sequences, which are commonly used for genome analysis. Comparison of the homologous SSRs among different genomes is helpful to understand the evolutionary process in relative species. In this study, SSR scanning was performed to investigate their distribution and length variation among the genomes of(D5),(A2) and(AD1). The results demonstrated that the distribution of SSRs in A genome was very similar with that in D genome, while the length variation of homologous SSRs between A and AD genome was more conserved than that between D and AD genome. Compared with SSRs in AD genome, the number of SSRs with longer motif length in A genome was about five times of those with shorter motif length, while it was about three times in D genome. This implied that the length variation rates of homologous SSRs between diploid cotton and tetraploid cotton were different during the parallel evolution due to the subgenome fusion, and the motif length of most SSRs in tetraoploid genome tended to become shorter than homologous SSRs in diploid genome during the process of evolution. This study comprehensively compared the SSRs in three cotton genomes and revealed the significant difference among them, providing a foundation for further evolutionary study ofgenome.
SSR; cotton genome; homologous SSR; evolution
2014-08-15;
2014-09-24
“十二五”国家支撑计划项目(编号:2013BAD01B03)和科技部、财政部国家科技基础条件平台项目(编号:2012-014)资助
孙高飞,博士,副教授,研究方向:棉花生物信息学。E-mail: sungaofei@sina.com何守朴,硕士,助理研究员,研究方向:棉花种质资源学。E-mail: zephyr0911@126.com孙高飞和何守朴并列第一作者。
杜雄明,博士,研究员,研究方向:棉花种质资源学。E-mail: dujeffrey8848@hotmail.com
10.16288/j.yczz.14-274
网络出版时间: 2014-12-15 8:49:34
URL: http://www.cnki.net/kcms/detail/11.1913.R.20141215.0849.001.html
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
新医学(2023年10期)2023-12-09 15:04:51
华人时刊(2023年1期)2023-03-14 06:43:36
南方医科大学学报(2022年3期)2022-04-13 01:51:26
汉字汉语研究(2021年2期)2021-08-30 08:58:46
今日农业(2021年11期)2021-08-13 08:53:24
浙江大学学报(农业与生命科学版)(2021年3期)2021-07-10 07:07:36
河北书画研究(2016年3期)2016-04-28 08:55:35
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
