
以PLK1 PBD为靶点的小分子抑制剂筛选及其抗癌活性研究
2015-11-25 01:18:12张智慧张晶刘伟陈云雨司书毅王艳宏
中国医药生物技术 2015年2期
张智慧,张晶,刘伟,陈云雨,司书毅,王艳宏
以PLK1 PBD为靶点的小分子抑制剂筛选及其抗癌活性研究
张智慧*,张晶*,刘伟,陈云雨,司书毅,王艳宏
目的 利用荧光偏振模型进行 PLK1 PBD抑制剂的高通量筛选,并对阳性化合 6143D7的抗癌效果进行体外评价。
方法 采用噻唑蓝比色法检测初筛阳性化合物对不同细胞系增殖的影响;流式细胞术检测化合物对细胞周期以及凋亡率的影响;分子对接检测化合物与 PLK1 PBD结构域的亲和性。
结果 利用荧光偏振模型得到的阳性化合物 6143D7经噻唑蓝比色法检测显示能抑制癌细胞的增殖;流式细胞仪检测显示该化合物加药组 G2/M 期细胞比例高于对照组并能促进细胞凋亡;分子对接结果表明化合物与 PLK1 PBD结构域的亲和性良好。
结论 该化合物通过抑制 PLK1 PBD结构域的活性发挥抗癌作用,有望成为靶向 PLK1的抗肿瘤先导化合物。
蛋白质丝氨酸苏氨酸激酶; 药物筛选试验,抗肿瘤; 荧光偏振; polo样激酶 1抑制剂
激酶是调控正常细胞和肿瘤细胞增殖的主要蛋白,polo样激酶(polo-like kinases,PLKs)是一类结构和功能均高度保守的丝/苏氨酸蛋白激酶[1-2],已经报道的PLKs(PLK1、PLK2、PLK3、PLK4)在结构方面具有很大的相似性,它们的 N端具有一个高度同源的激酶催化结构域[3-4],C端具有调节 PLKs催化活性及亚细胞动态定位的特征性结构域(polo-box domain,PBD)[5]。PLK家族不同成员的 PBD对同一底物的亲和力不同,PBD1有两个特异的 polo-box(PB1和 PB2),PB1由 78个氨基酸组成,PB2序列也相差不大,而 PBD4为两个 PLK4聚合后的变异体,C端序列存在很大的特异性,且仅存在一个 polo-box[6]。
PLK1主要在有丝分裂的 G2晚期和 M 期表达,参与有丝分裂的多个阶段,在四种激酶中最为重要[7],被公认为是一个潜在的治疗癌症药物的靶点[8-9]。然而,目前大部分抗 PLK1小分子抑制剂均是针对激酶结构域的 ATP结合口袋(ATP-binding pocket)进行设计的[10-11],但 ATP结合区的结构保守性使其对其他激酶的抑制作用难以区别,并且还容易与类似的药物有交叉耐药性。所以,为寻求更高效、专一的 PLK1抑制剂,PBD1成为了近年来寻找高选择性 PLK1抑制剂的新靶点[12-19]。
荧光偏振方法是 Perrin[20]于 1926年提出的理论,通过检测荧光标记的分子或者固有荧光分子的荧光强弱变化来研究分子间相互作用,能够直接、近乎瞬时地检测结合于自由示踪剂分子的比值。当小的荧光分子在平面极化光的激发下,荧光寿命内(激发与发射之间的时差)小分子在溶液中的旋转速度快,所形成的发射光大部分是去极化的;但是,当荧光分子结合到大分子后,它的有效体积增大,旋转减慢,在与激发光相同平面上的发射光增多,去极化减弱,偏振增强。因此,荧光分子在自由状态下偏振值较低,而在结合状态偏振值较高,检测到的偏振值是这两个值的加权平均值,从而提供了直接检测荧光分子结合到受体比值的方法[21-24]。这种技术被广泛应用于临床和生物医学领域,包括某些疾病的诊断和监测治疗药物在体内的水平,进而用于高通量筛选,为药物的筛选提供了更加灵敏、简易、快速的方法。本实验利用荧光偏振法对 PBD1抑制剂进行筛选,并进行一系列的活性验证。
1 材料与方法
1.1 材料
1.1.1 菌株 人肺腺癌细胞 A549、人宫颈癌细胞HeLa、人大细胞肺癌细胞 H460、人肾脏细胞Herk-293、人巨噬细胞 Raw、人直肠癌细胞 HT29、人结肠癌细胞 HCT116及人前列腺癌细胞 PC3均由中国医学科学院医药生物技术研究所重点实验室保存。
1.1.2 试剂 AnnexinV-FITC/PI双染试剂盒购自北京四正柏生物科技有限公司;10 000个待筛化合物购自北京百灵威科技有限公司;PBD1重组蛋白由中国医学科学院医药生物技术研究所重点实验室陈云雨博士提供;DMEM高糖培养基(hyclone)购自美国 Sigma公司;30%丙烯酰胺、4×浓缩胶缓冲液、4×分离胶缓冲液、考马斯亮蓝染色、NaCl、Tris-HCl、EDTA、RNA酶A(RNaseA)、碘化丙啶(PI)、噻唑蓝(MTT)、DMSO均购自北京普利莱基因技术有限公司。
1.1.3 主要仪器 Envision 2014 Multilabel Reader酶标仪购自美国 PerkinElmer公司;FACS Caliur流式细胞仪购自美国 Beckman Coulter公司;PowerPacTMHC稳压稳流电泳仪购自美国 Bio-Rad公司。
1.2 方法
1.2.1 荧光偏振法初筛化合物 30 μl体外表达纯化的 PBD1蛋白(400 nmol/L)中加入受试化合物 0.3 μl(1 mg/ml),于 25℃、140 r/min条件下振荡培养 45 min,再加入 FITC标记的多肽 30 μl (60 nmol/L),于 25℃、140 r/min下避光振荡培养 15 min,酶标仪检测。其中只加 PBD1和 FITC标记多肽的作为阳性对照,只加缓冲液和多肽的作为阴性对照。抑制率计算公式如下:

1.2.2 初筛阳性化合物 IC50值的确定 分别将化合物稀释为 10、5、2、1、0.5、0.25、0.1、0.05、0.02、0.01 μg/ml。按初筛的步骤进行实验,应用GraphPad Prism 5计算化合物在此荧光偏振模型上的半数抑制浓度(IC50)。
1.2.3 MTT法检测化合物细胞生长抑制率 取对数生长期细胞,调整细胞数为 2×104~5× 104/ml,接种于 96孔培养板中。细胞贴壁后加入浓度梯度稀释的化合物,化合物浓度分别为 200、100、75、25、10、5、1、0.5、0.1 μmol/L,同一浓度设 3个复孔。于 37℃ 含 5%CO2的培养箱中培养 48 h,然后每孔加入 20 μl MTT(5 mg/ml)继续孵育 4 h,再加入 150 μl DMSO振荡 15 min。酶联免疫检测仪测定 570 nm波长处 OD值。

应用 GraphPad Prism 5计算化合物对细胞生长抑制的半数抑制浓度(IC50)。
1.2.4 细胞周期检测 将 HeLa细胞铺于 6孔板中,待细胞贴壁后加入 40 μl胸腺嘧啶(0.1 mol/L),使其终浓度为 2 mmol/L,此过程为第一次阻断,孵箱培养 8~12 h后,PBS洗 3次,加入正常培养基培养 12 h后进行第二次阻断,条件同第一次,经两次阻断后加入浓度分别为 5、10、15、20、25 μmol/L的化合物 6143D7。收集作用 16 h后的样品,PBS洗涤,以预冷的 70%乙醇固定至少1h。PBS洗涤、重悬细胞,加入 RNaseA,37℃ 恒温孵育 30 min,加 PI室温避光孵育 30 min,1 h内流式细胞仪检测,以 ModFitLT软件分析结果。
1.2.5 Annexin V-FITC/PI双染法检测细胞凋亡 收集 6143D7(0、15、25 μmol/L)作用 24 h后的 HeLa细胞,以 4℃ 预冷的 PBS洗涤,结合缓冲液重悬浮细胞,调节细胞数为 1×106个/ml,加入 Annexin V-FITC后室温避光孵育 30 min,上机前 5 min加入 PI,FACS分析。
1.2.6 化合物 6143D7与 PLK1激酶 PBD结构域分子对接 利用 SYBYL7.3软件首先将蛋白调出并对其进行 3D模型的优化,确定活性位点,再将化合物与活性位点进行对接。
2 结果
2.1 荧光偏振模型初筛结果
利用荧光偏振模型对 10 000个化合物进行PBD1抑制剂初筛,得到 1个阳性化合物6143D7,在浓度为 1 mg/ml时能抑制 PBD1的活性,化合物结构如图 1所示。

图1 化合物 6143D7的结构式Figure 1 The structure of compound 6143D7
2.2 初筛阳性化合物对 PBD1的抑制活性
对初筛阳性化合物采用浓度梯度稀释进行 IC50的测定,发现化合物 6143D7表现出较好的抑制作用,IC50值为(0.143±0.022)μmol/L(图 2)。

图2 6143D7对体外纯化 PBD1的 IC50值Figure 2 The IC50of 6143D7 against purified PBD1 in vitro
2.3 MTT法检测化合物对细胞的生长抑制
分别以不同细胞系进行 MTT实验,发现在A549、H460、HCT116、HT29上化合物 6143D7的作用并不敏感,对 HeLa细胞作用敏感,IC50= 5.78 μmol/L,Raw细胞中 IC50值为 50.76 μmol/L,如表 1和图 3所示。6143D7对 HeLa细胞敏感,且 HeLa细胞比较常用,故后续实验均采用HeLa细胞进行。

表1 6143D7对不同细胞株的毒性Table 1 Cytotoxicity of 6143D7 to different cells
2.4 化合物对细胞周期的影响
由于在细胞培养过程中,不同时相的细胞对药物干预存在不同反应,会影响实验的重复性,因此,需要获得周期一致性的细胞,利用细胞同步化技术可以达到此效果。将 HeLa细胞进行胸腺嘧啶核酸双阻断,以 6143D7作用 16 h,流式细胞仪检测发现 G2/M 期随着化合物浓度的增加在整个细胞周期所占比例也有所增加。尤其是在 25 μmol/L时,由对照组的 0%增加到 22.33%(表 2)。这表明 6143D7有 G2/M 期阻滞作用,并且这种作用有明显的剂量依赖关系。
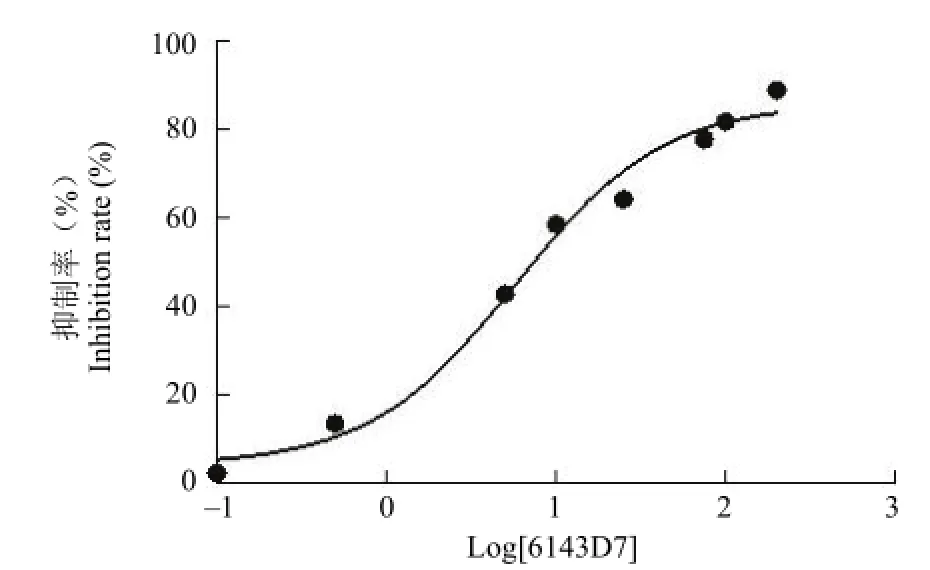
图3 6143D7在 HeLa细胞中的半数抑制率Figure 3 The IC50of 6143D7 for HeLa cells
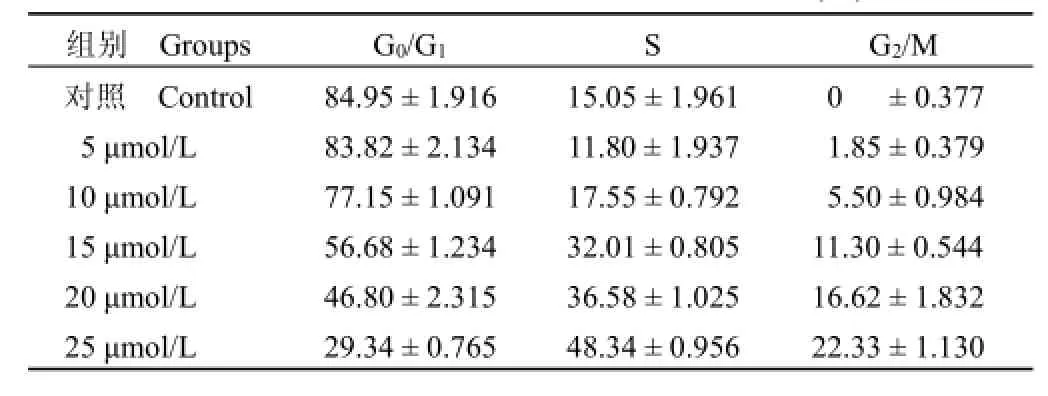
表2 不同浓度的 6143D7作用下HeLa细胞周期分布(%)Table 2 Effect of 6143D7 on cell cycle progression of HeLa cells at different concentrations(%)
2.5 Annexin V-FITC/PI双染法检测细胞凋亡
Annexin V-FITC/PI双染法结果显示,6143D7能够促进 HeLa细胞的凋亡,尤其是晚期凋亡。如图 4及表 3所示,6143D7作用 24 h后,15 μmol/L的化合物能诱导 22.69%的细胞发生晚期凋亡,25 μmol/L的化合物能诱导 79.15% 的细胞发生晚期凋亡。
2.6 化合物与 PLK1 PBD分子对接
PLK1 PBD结构域由 Lys540、His538、Lys540、Tyr485、Arg516、Asp416等氨基酸组成[25-30]。对接结果显示,化合物 6143D7与 PLK1的 PBD结构域呈现出了较高的亲和性,如图 5的球棒模型图所示,化合物 6143D7可以进入 PBD1的活性口袋中,且对接方式主要是以 Lys540与苯环上羰基形成的氢键为主。
3 讨论
近年来,癌症的发生一直是危害人类健康的重要因素,各国科研人员也不断地致力于抗癌药物的研究。PLK1作为抗癌药物的研究靶点,由于PLKs之间具有相似的激酶区结构,使得这些抑制剂或多或少地对 PLK2及 PLK3也有抑制作用,因此,其特异性就受到了限制。然而,PBD的特殊结构成为寻找特异性 PLK1小分子抑制剂的一个新领域。
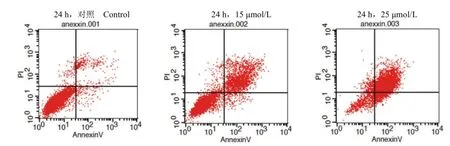
图4 6143D7对 HeLa细胞的促凋亡作用Figure 4 Treatment of HeLa cells with compound 6143D7 induced apoptosis

表3 不同浓度的 6143D7作用下 HeLa细胞凋亡分布(%)Table 3 Effect of 6143D7 on cell apoptosis of HeLa cells at different concentration(%)
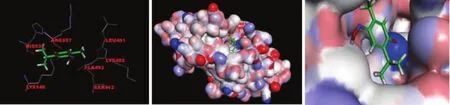
图5 6143D7与 PBD1活性区域的虚拟对接Figure 5 Virtual docking of 6143D7 with PBD1 domain
目前,已经报道的 PLK1 PBD抑制剂主要包括小分子抑制剂和多肽类抑制剂。小分子抑制剂主要包括 poloxin[30]及其结构类似物百里醌(thymoquinone,TQ)[31]、poloxipan、红倍酚(purpurogallin,PPG)和绿茶儿茶素类等。但是,这些数量及结构有限的小分子抑制剂多属天然产物,极大地限制了靶向 PLK1 PBD小分子抑制剂基于结构的定向设计。鉴于此,本研究采用荧光偏振高通量筛选模型对本室化合物库进行高通量筛选,以期定向获得靶向 PLK1 PBD的新型抗肿瘤药物先导化合物,初筛得到的化合物 6143D7是一类结构新型的抗肿瘤化合物,MTT实验发现6143D7在 HeLa细胞中抑制作用最为明显,可能和此化合物作用的靶点蛋白在这几种细胞系中的表达量不同有关,这也就决定了其对细胞系的不同敏感性。流式结果还表明,6143D7对癌细胞系的增殖抑制作用可能是通过诱导细胞周期阻滞和促进细胞凋亡来实现的。6143D7导致 HeLa细胞G2/M 期阻滞呈一定的量效关系,在 25 μmol/L 6143D7作用下,细胞 G2/M 期的比例明显高于对照组。6143D7促进 HeLa细胞的凋亡则更为明显,量效关系也更突出,这也与已发现的 PBD1抑制剂作用效果类似,经过分子对接也进一步证实了PBD1为化合物的作用靶点。
总之,本研究验证了 6143D7的体外抗癌活性,提示其具有较好的抗癌研究和开发前景,表明该化合物具有进一步研究的价值。接下来将对化合物进行结构改造以进一步提高其抗肿瘤活性。
[1] Fenton B,Glover DM.A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos.Nature,1993,363(6430):637-640.
[2] Glover DM,Hagan IM,Tavares AA.Polo-like kinaes:a team that plays thoughout mitosis.Genes Dev,1998,12(24):3777-3787.
[3] Kothe M,Kohls D,Low S,et al.Structure of the catalytic domain of human polo-like kinase 1.Biochemistry,2007,46(20):5960-5971.
[4] Strebhardt K.Multifaceted polo-like kinases:drug targets and antitargets for cancer therapy.Nat Rev Drug Discov,2010,9(8):643-660.
[5] de Cárcer G,Manning G,Malumbres M.From Plk1 to Plk5: functional evolution of polo-like kinases.Cell Cycle,2011,10(14):2255-2262.
[6] Zhang L,Cao YH,Lu S,et al.Targeting the substeate binding domain of polo-like kinase 1:advances in the study of PBD1 inhibitors.Acta Pharm Sinica,48(3):315-324.(in Chinese)
张亮,曹燕华,卢帅,等.靶向Polo样激酶1底物结合区:PBD1抑制剂的研究进展.药学学报,2013,48(3):315-324.
[7] Park JE,Sooung NK,Johmura Y,et al.Polo-box domain:a versatile mediator of polo-like kinase function.Cell Mol Life Sci,2010,67(12): 1957-1970.
[8] Lowery DM,Mohammad DH,Elia AE,et al.The Polo-box domain:a meolecular integrator of mitiotic kinase cascades and Polo-like function.Cell Cycle,2004,3(2):128-131.
[9] Rudolph D,Steegmaier M,Hoffmann M,et al.BI 6727,a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity.Clin Cancer Res,2009,15(9):3094-3102.
[10]Goh KC,Wang H,Yu N,et al.PLK1 as a potential drug target in cancer therapy.Drug Dev Res,2004,62(4):349-361.
[11]Schöffski P,Blay JY,De Greve J,et al.Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer,breast cancer,ovarian cancer,soft tissue sarcoma and melanoma.The first protocol of the European Organization for Research and Treatment of Cancer(EORTC)Network of Core Institutes(NOCI).Eur J Cancer,2010,46(12):2206-2215.
[12]Lee KS,Idle JR.Pinning down the polo-box domain.Chem Biol,2008,15(5):415-416.
[13]Strebhardt K.Multifaceted polo-like kinases:drug targets and antitargets for cancer therapy.Nat Rev Drug Discov,2010,9(8):643-660.
[14]Yun SM,Moulaei T,Lim D,et al.Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1.Nat Struct Mol Biol,2009,16(8):876-882.
[15]Liu F,Park JE,Qian WJ,et al.Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel.Nat Chem Biol,2011,7(9):595-601.
[16]Richter S,Neundorf I,Loebner K,et al.Phosphopeptides with improved cellular uptake properties as ligands for the polo-box domain of polo-like kinase 1.Bioorg Med Chem Lett,2011, 21(16):4686-4689.
[17]Śledź P,Stubbs CJ,Lang S,et al.From crystal packing to molecular recognition:prediction and discovery of a binding site on the surface of polo-like kinase 1.Angew Chem Int Ed Engl,2011,50(17): 4003-4006.
[18]Śledź P,Lang S,Stubbs CJ,et al.High-throughput interrogation of ligand binding mode using a fluorescence-based assay.Angew Chem Int Ed Engl,2012,51(31):7680-7683.
[19]Qian WJ,Park JE,Lim D,et al.Peptide-based inhibitors of Plk1 polo-box domain containing mono-anionic phosphothreonine esters and their pivaloyloxymethyl prodrugs.Chem Biol,2013,20(10):1255-1264.
[20]Perrin F.Polarization of light of fluorescence,average life of molecules in the excited state.J Phys Radium,1926,7(12):390-401.
[21]OwickiJC.Fluorescence polarization and anisotropy in high throughput screening:perspectives and primer.J Biomol Screen,2000,5(5):297-306.
[22]Burke TJ,Loniello KR,Beebe JA,et al.Development and application of fluorescence polarization assays in drug discovery.Comb Chem High Throughput Screen,2003,6(3):183-194.
[23]Jameson DM,Croney JC.Fluorescence polarization:past,present and future.Comb Chem High Throughput Screen,2003,6(3):167-173.
[24]Nasir MS,Jolley ME.Fluorescence polarization:an analytical tool for immunoassay and drug discovery.Comb Chem High Throughput Screen,1999,2(4):177-190.
[25]Emmitte KA,Adjabeng GM,Andrews CW,et al.Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding.Bioorg Med Chem Lett,2009,19(6): 1694-1697.
[26]Reindl W,Yuan J,Krämer A,et al.Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol,2008,15(5):459-466.
[27]Reindl W,Yuan J,Krämer A,et al.A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem,2009,10(7):1145-1148.
[28]Murugan RN,Park JE,Lim D,et al.Development of cyclic peptomer inhibitors targeting the polo-box domain of polo-like kinase 1.Bioorg Med Chem,2013,21(9):2623-2634.
[29]Cheng KY,Lowe ED,Sinclair J,et al.The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex.EMBO J,2003,22(21):5757-5768.
[30]Reindl W,Yuan J,Krämer A,et al.Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol,2008,15(5):459-466.
[31]YinZ,Song Y,RehsePH.ThymoquinoneblockspSer/pThr recognition by Plk1 Polo-box domain as a phosphate mimic.ACS Chem Biol,2013,8(2):303-308.
Methods The influence of the positive compound on cells'proliferation was measured by MTT assay.The compound's effects oncell cycle and apoptosis in HeLa cells was decided by flow cytometry(FCM).The compound’s affinity with PBD1 domain was determined by molecular docking.
Results The positive compound 6143D7 was found from high-throughput screening by fluorescence polarization assay.In vitro antitumor results revealed that the compound inhibited the proliferation of human cancer cell lines.6143D7 induced cell cycle arrest and promoted apoptosis of the cancer cells by FCM assess.Molecular docking showed that 6143D7 had good affinity with the PBD1 domain.
Conclusion The compound 6143D7 is expected to become one of the anti-cancer drug candidates against PLK1 PBD.
Screening and anti-tumor activity of small molecular inhibitors targeting PLK1 PBD
ZHANG Zhi-hui,ZHANG Jing,LIU Wei,CHEN Yun-yu,SI Shu-yi,WANG Yan-hong
Objective Fluorescence polarization assay will be used to find potential small-molecular inhibitors targeting PLK1 PBD through high-throughput screening,and then the anti-tumor effects of positive compound 6143D7 will be evaluated in vitro.
Protein-serine-threonine kinases;Drug screening assays,antitumor;Fluorescence polarization;Polo-like kinase 1 inhibitor
s:SI Shu-yi,Email:sisyimb@hotmail.com;WANG Yan-hong,Email:wang.yanhong@163.com
10.3969/cmba.j.issn.1673-713X.2015.02.006
国家自然科学基金面上项目(81370087)
150040哈尔滨,黑龙江中医药大学药学院(张智慧、王艳宏);100050北京,中国医学科学院医药生物技术研究所国家新药(微生物)筛选实验室(张晶、刘伟、陈云雨、司书毅)
司书毅,Email:sisyimb@hotmail.com;王艳宏,Email:wang.yanhong@163.com
2015-01-07
*同为第一作者
www.cmbp.net.cn 中国医药生物技术,2015,10(2):125-130
Author Affiliations:College of Pharmacy,Heilongjiang University of Chinese Medicine,Harbin 150040,China(ZHANG Zhi-hui,WANG Yan-hong);National Laboratory for Screening New(Microbial)Drugs,Institute of Medicinal Biotechnology,Academy of Medical Sciences&Peking Union Medical College,Beijing 100050,China(ZHANG Jing,LIU Wei,CHEN Yun-yu,SI Shu-yi)
www.cmbp.net.cn Chin Med Biotechnol,2015,10(2):125-130
猜你喜欢
科学导报(2024年19期)2024-04-22 05:53:32
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
中华养生保健(2020年3期)2020-11-16 00:52:28
电子制作(2019年12期)2019-07-16 08:45:20
World Journal of Clinical Cases(2019年6期)2019-04-17 02:25:08
测控技术(2018年6期)2018-11-25 09:50:24
山西大同大学学报(自然科学版)(2016年2期)2016-12-12 03:19:15
中国卫生标准管理(2015年3期)2016-01-14 03:41:43
中国医药生物技术(2015年4期)2015-12-26 08:26:36
