
食用林产品中氨基甲酸酯类农药残留量测定
2015-11-18 02:24杨振兴邓洁红
湖南林业科技 2015年1期
杨振兴, 邓洁红, 席 慧
(1.湖南农业大学食品科技学院, 湖南 长沙 410004;2.长沙环境保护职业技术学院, 湖南 长沙 410004)
食用林产品中氨基甲酸酯类农药残留量测定
杨振兴1,2, 邓洁红1*, 席 慧2
(1.湖南农业大学食品科技学院, 湖南 长沙 410004;2.长沙环境保护职业技术学院, 湖南 长沙 410004)
建立了分散固相萃取-高效液相色谱-串联质谱联用法测定食用林产品(香菇)中14种氨基甲酸酯类农药残留量的方法。样品经乙腈提取,PSA和少量GCB混合吸附剂分散固相萃取净化,然后采用HPLC-MS/MS测定,外标法定量。14种氨基甲酸酯类农药在0.005~0.100 0 mg/L范围内线性良好,相关系数均大于0.995 8。在0.01~0.05 mg/kg浓度范围内,14种目标物的平均加标回收率为70.0%~104.5%,相对标准偏差为3.2%~9.1%。该方法简单、快速、灵敏,可满足香菇中14种氨基甲酸酯类农药残留的检测需要。
氨基甲酸酯类; 农药残留; 林产品; 高效液相色谱-串联质谱
食用林产品是指食用菌、板栗、猕猴桃等可供人食用的林产品[1-2]。随着林业经济的快速发展,可产生巨大经济价值的食用林产品的人工林种植面积也不断扩大[3],为了保证种植的产物能够有效地避免害虫危害,在种植过程中就不得不施用大量农药,农药残留问题也就难以避免了,从而给人类带来了很大的危害[4-6]。目前,氨基甲酸酯类农药凭借其高效、广谱、分解快、残留时间短的特点,逐渐在食用林产品生产中使用得越来越广,其残留造成的食用林产品安全问题也引起了人们的广泛关注。因此,国际上对食品中氨基甲酸酯提出了严格的限量,从而使优势食用林产品的出口遭受了技术壁垒[7]。只有不断地提高分析检测能力,建立快速、高效的分析方法,才能扩大我国食用林产品的竞争力,保障人们的食用安全。
目前国内外用于食品中氨基甲酸酯类农药残留检测的方法主要有气相色谱法、气质联用法、高效液相色谱法、液相串联质谱法等[8-15]。分散固相萃取法是近些年在食品农药残留检测中越来越受欢迎的一种样品前处理技术,该方法相对传统的前处理方法具有使用有机试剂少、快速、简便、成本低等优点,并且能够有效地除去样品基质中色素、有机酸等干扰物的干扰,完全能够满足出口食用林产品大批量检测的需要。我们以食用林产品香菇为研究对象,采用PSA和GCB为分散固相萃取的吸附剂,结合液相色谱-串联质谱分析技术实现食用林产品中多种氨基甲酸酯类农药残留的快速、有效地检测。
1 材料与方法
1.1 材料、仪器与试剂
Agilent 1200高效液相色谱仪(美国安捷伦公司);API4000型三重四极杆串联质谱仪(美国AB公司);SK-1快速混匀器(常州澳华);2-16P离心机(德国SIGMA公司)。
灭害威、涕灭威亚砜、涕灭威砜、灭多威、久效威、涕灭威、恶虫威、残杀威、克百威、乙硫苯威、异丙威、仲丁威、甲硫威、猛杀威等氨基甲酸酯类农药标准品均购于德国Dr.Eh- renstorfer公司,纯度大于97.5%。乙腈、甲醇、乙酸为色谱纯;无水硫酸钠、无水硫酸镁为分析纯;PSA粉末、GCB粉末购于美国Supelco公司;水为超纯水。香菇购于当地市场。
1.2 试验方法
1.2.1 混合标准溶液的配制 分别称取适量的农药标准物置于10 mL容量瓶中,用乙腈定容至刻度,配制成浓度为 1 mg/mL 的标准储备液,于-4 ℃冰箱中避光冷冻保存。然后移取一定量的各单标储备液到另一10 mL容量瓶,用乙腈定容至刻度,配制成10 μg/mL的混合标准溶液,按照检测需要用乙腈稀释成一系列标准工作溶液。
1.2.2 样品的前处理 准确称取粉碎均匀的香菇样品5.0 g(精确到0.01 g)于50 mL具塞刻度离心管中,加入10 mL乙腈、2 g氯化钠,涡旋振荡3 min,以6 000 r/min离心5 min,取上层清液到另一洁净的刻度离心管中,下层残渣再用10 mL乙腈按前面的步骤提取一次,离心分离后,合并两次的提取溶液。取4 mL上清液到15 mL洁净的具塞刻度离心管中,加入100 mg PSA、10 mg GCB、1 g 无水硫酸钠和1 g无水硫酸镁,涡旋振荡1 min,以6 000 r/min离心5 min后,取1 mL上清液过0.45 μm有机滤膜,待上机分析。
1.2.3 样品的测定
(1)液相色谱条件
色谱柱:Agilent C18柱(5 μm,150 mm×4.6 mm);流动相A:0.1%乙酸,流动相B:乙腈,梯度洗脱程序:0~5 min,10% B~40% B;5~10 min,50% B~90% B;10~17 min,90% B;17~18 min,90% B~10% B;18~20 min,10%B。流速:0.5 mL/min;柱温:35 ℃;进样量:10 μL。
(2)质谱条件
电喷雾离子源;正离子扫描方式,多反应监测(MRM),电喷雾电压:5 000 V,离子源温度:500 ℃;雾化气流速:310 kPa;辅助加热气:345 kPa;帘气流速:70 kPa;碰撞气流速:50 kPa;14种氨基甲酸酯农药的质谱分析参数见表1。
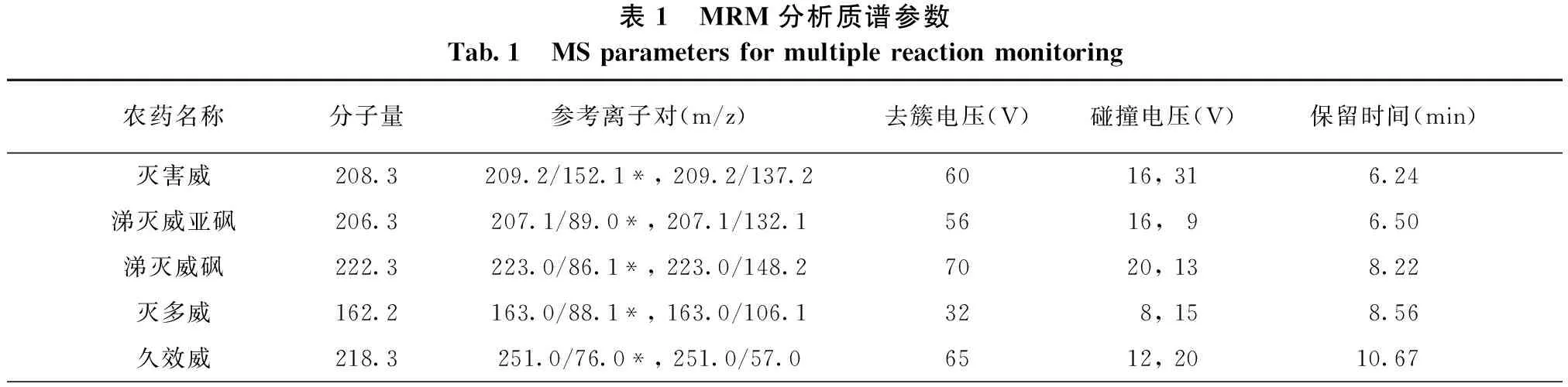
表1 MRM分析质谱参数Tab.1 MSparametersformultiplereactionmonitoring农药名称分子量参考离子对(m/z)去簇电压(V)碰撞电压(V)保留时间(min)灭害威208.3209.2/152.1*,209.2/137.26016,316.24涕灭威亚砜206.3207.1/89.0*,207.1/132.15616,96.50涕灭威砜222.3223.0/86.1*,223.0/148.27020,138.22灭多威162.2163.0/88.1*,163.0/106.1328,158.56久效威218.3251.0/76.0*,251.0/57.06512,2010.67
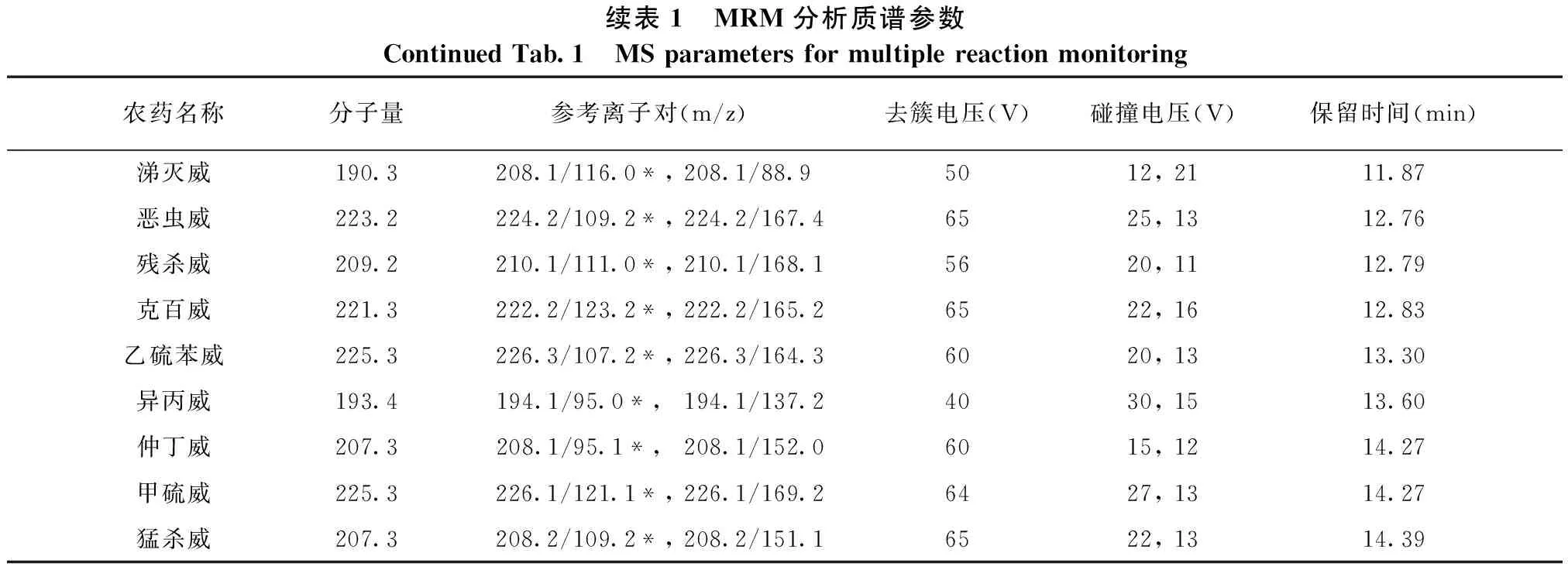
续表1 MRM分析质谱参数ContinuedTab.1 MSparametersformultiplereactionmonitoring农药名称分子量参考离子对(m/z)去簇电压(V)碰撞电压(V)保留时间(min)涕灭威190.3208.1/116.0*,208.1/88.95012,2111.87恶虫威223.2224.2/109.2*,224.2/167.46525,1312.76残杀威209.2210.1/111.0*,210.1/168.15620,1112.79克百威221.3222.2/123.2*,222.2/165.26522,1612.83乙硫苯威225.3226.3/107.2*,226.3/164.36020,1313.30异丙威193.4194.1/95.0*, 194.1/137.24030,1513.60仲丁威207.3208.1/95.1*, 208.1/152.06015,1214.27甲硫威225.3226.1/121.1*,226.1/169.26427,1314.27猛杀威207.3208.2/109.2*,208.2/151.16522,1314.39
2 结果与分析
2.1 提取溶剂的选择
选取甲醇、乙腈、二氯甲烷和丙酮作为提取溶剂进行比较,结果表明(见图1),采用乙腈和丙酮提取的回收率均高于甲醇和二氯甲烷提取的回收率,且二氯甲烷提取过程中易发生乳化现象。另外采用丙酮提取时,提取出来的杂质稍多,对谱图有一定的干扰,在进液质分析前需要转换溶剂,过程稍微复杂一点。采用乙腈提取对14种氨基甲酸酯类农药均可以获得较满意的回收率,并且操作过程相对简便,因此实验最终选取乙腈作为提取溶剂。
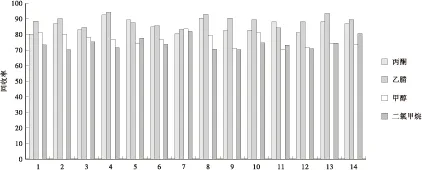
1.灭害威;2.涕灭威亚砜;3.涕灭威砜;4.灭多威;5.久效威;6.涕灭威;7.恶虫威;8.残杀威;9.克百威;10.乙硫苯威;11.异丙威;12.仲丁威;13.甲硫威;14.猛杀威图1 不同提取溶剂对14种氨基甲酸酯类农药回收率的影响Fig.1 Effect of different extract solvents on the recoveries of 14 carbamate pesticide
2.2 提取溶剂用量的优化
提取溶剂的用量也是影响待测物回收率的一个重要因素,在保证农药回收率的基础上尽量减少提取溶剂的用量,一方面可以降低成本,一方面可以减少废液对环境的污染。实验考察了10、20、30、40 mL四种不同用量的乙腈对5 g香菇样品(添加浓度为0.1 mg/kg)中待测目标物的提取效率。结果表明,提取溶剂较少时,不能完全将目标化合物从样品基质中提取出来,使用较多的提取溶剂不仅浪费,还需要消耗较多的离心分离时间。当乙腈用量为20 mL时,14种氨基甲酸酯类农药的平均回收率均大于90%,能够满足检测的需要,因此实验确定样品的提取溶剂用量为20 mL,分两次进行提取。
2.3 吸附剂的选择
在实验过程中发现,吸附剂的类型对样品提取溶液的净化效果以及目标化合物的萃取效率有着非常大的影响。实验比较了分散固相萃取常用的几种吸附剂如PSA、C18和ODS的吸附净化效果。由于香菇样品中存在色素、有机酸等具有一定极性的干扰物质,C18和ODS吸附剂的净化效果相对较差,去除这些杂质的能力较弱。PSA则凭借离子交换作用能有效地吸附色素、有机酸这些干扰物质,进而达到对样品提取液的净化目的。由于香菇提取溶液的颜色稍深,还可考虑加入少量的GCB来进一步去除色素。实验还比较了当PSA用量为100 mg,GCB用量分别为5、10、20和40 mg时,对14种氨基甲酸酯类农药的回收率影响,结果表明GCB的用量不宜过多,否则对目标化合物的吸附性过强,影响目标化合物的回收率。综合考虑去除色素效果和回收率,实验最终选取PSA用量为100 mg,GCB用量为10 mg的组合为实验的吸附剂。
2.4 测定条件的优化
2.4.1 液相色谱条件的优化 氨基甲酸酯类农药易溶于乙腈,并且该类农药在酸性条件下更容易促进其电离,增强响应值,故考察了不同乙酸含量-乙腈的流动相体系,结果表明,采用乙酸-乙腈体系作为流动相进行梯度洗脱时,乙酸促进正离子的形成,待测农药目标物灵敏度较高,但随着乙酸的比例不断增加,相对应的质谱基线噪音也随之增大,反而使得灵敏度降低。通过对比发现,0.1%乙酸-乙腈是最佳的流动相体系,能够有效地对色谱柱吸附的目标物进行洗脱,实现对待测物的准确定性和定量。
2.4.2 质谱条件的优化 优化的色谱条件后,在ESI正离子模式下进行全扫描,得到各个化合物的母离子,然后通过调节锥孔电压和电离电压,确定各待测化合物的子离子后,再对碰撞电压进行优化,使得待测物的响应值达到最大 。由于待测的目标化合物如恶虫威、克百威、残杀威等的化学结构和极性相似,因此在总离子流图上这些化合物的色谱峰很容易发生重叠,直观上很难区分开。但通过多反应监测模式对具有不同分子量的化合物进行分离测定,各个化合物的色谱峰可单独显示,不受其他化合物的干扰(见图2)。
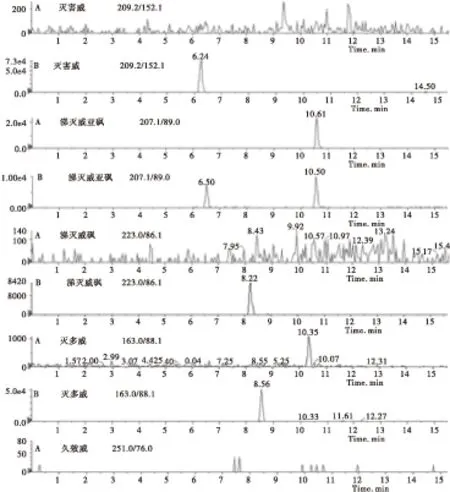
图2 香菇空白(A)和香菇样品添加14种氨基甲酸酯混合标准品(0.01 mg/L)的定量离子色谱图(B)Fig.2 Quantitative ion chromatograms of(A)mushroom blank and(B)mushroom sample with a mixture of 14 carbamate pesticide standards(0.01 mg/L)
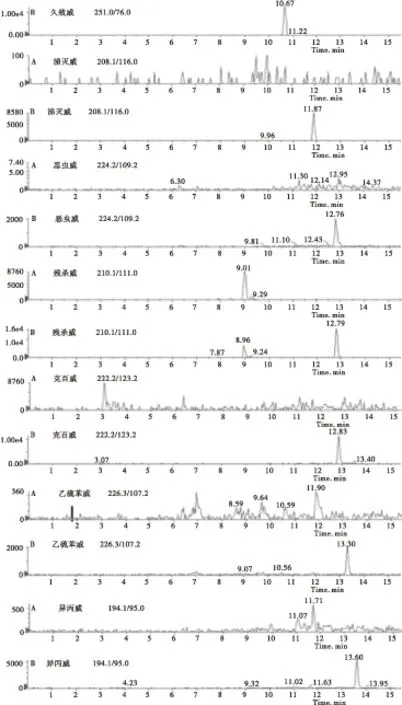
续图2 香菇空白(A)和香菇样品添加14种氨基甲酸酯混合标准品(0.01 mg/L)的定量离子色谱图(B)Continued Fig.2 Quantitative ion chromatograms of(A)mushroom blank and(B)mushroom sample with a mixture of 14 carbamate pesticide standards(0.01 mg/L)
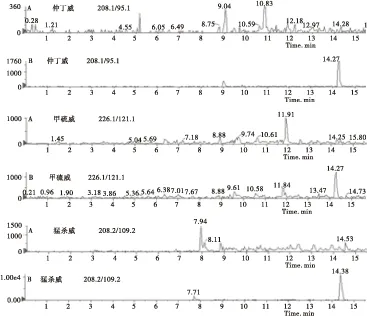
续图2 香菇空白(A)和香菇样品添加14种氨基甲酸酯混合标准品(0.01 mg/L)的定量离子色谱图(B)Continued Fig.2 Quantitative ion chromatograms of(A)mushroom blank and(B)mushroom sample with a mixture of 14 carbamate pesticide standards(0.01 mg/L)
2.5 线性关系和测定低限
在确定的最优实验条件下,移取一定量的混合标准溶液,用0.1%的乙酸:乙腈(1∶1,V/V)流动相稀释成一系列浓度逐渐递减的混合标准工作液进行测定。以混合工作液中各组分的仪器响应峰面积所对应的质量浓度绘制各组分的标准曲线。各组分的回归方程、相关系数和定量限如表2所示。14种氨基甲酸酯类农药在0.005~0.1 000 mg/L质量浓度范围内标准曲线线性关系良好,相关系数(r2)均大于0.995 8,定量限(LOQ)以实际测定时10倍性噪比(S/N)估算确定。
2.6 回收率和精密度
以典型的林产品香菇为基质,称取5.0 g样品,分别添加质量浓度为10 μg/mL的14种氨基甲酸酯类农药的混合标准溶液10 μL、25 μL、50 μL,每个添加浓度取6个平行样品,按照1.2.2中的方法步骤对样品进行前处理,进行三水平添加回收实验,回收率和精密度数据如表3所示。三个添加水平下的14种待测目标物回收率范围为70.0%~104.5%,相对标准偏差(RSD)为3.2%~9.1%。说明本方法完全可以满足农药残留的检测要求。
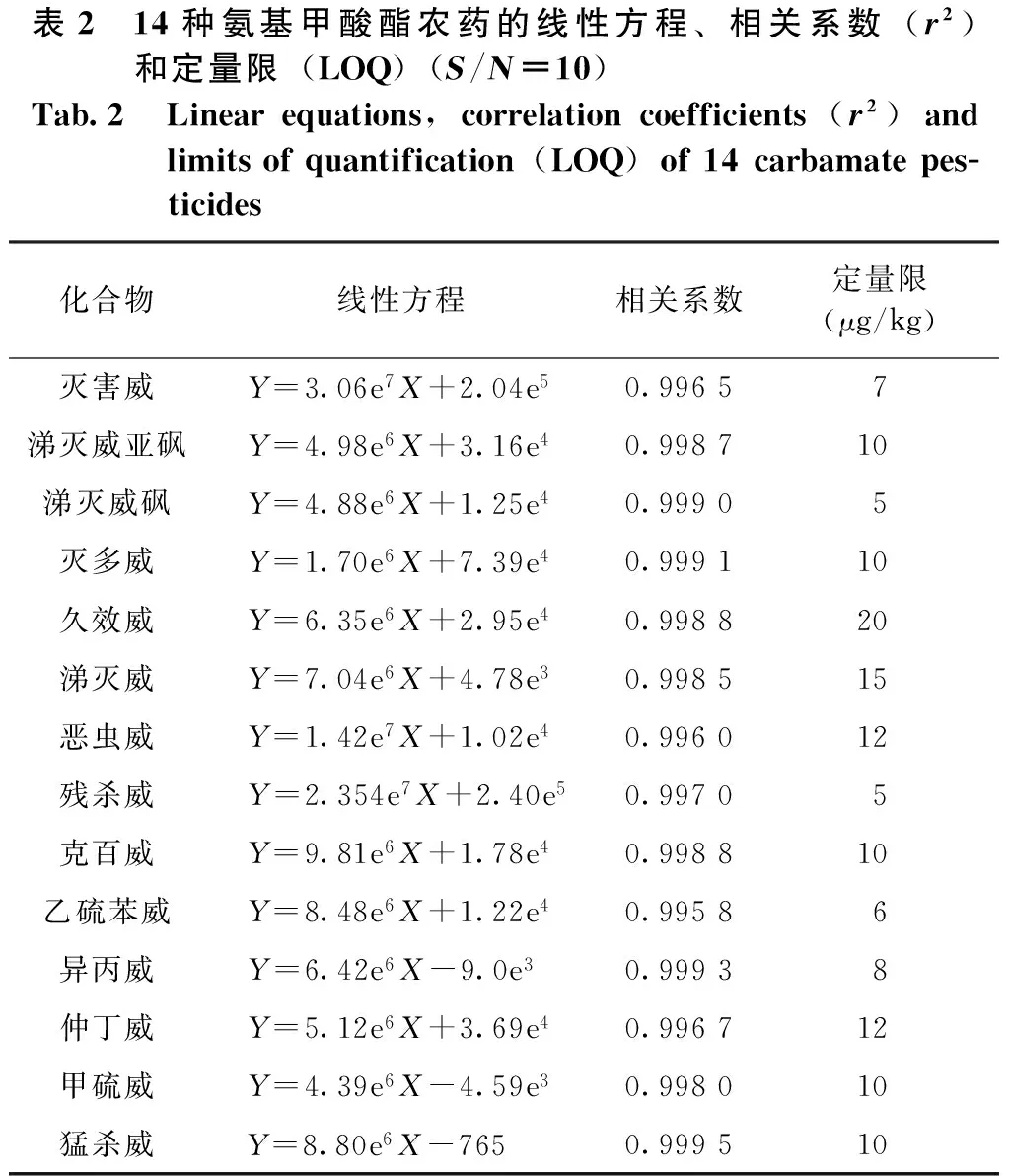
表2 14种氨基甲酸酯农药的线性方程、相关系数(r2)和定量限(LOQ)(S/N=10)Tab.2 Linearequations,correlationcoefficients(r2)andlimitsofquantification(LOQ)of14carbamatepes-ticides化合物线性方程相关系数定量限(μg/kg)灭害威Y=3.06e7X+2.04e50.99657涕灭威亚砜Y=4.98e6X+3.16e40.998710涕灭威砜Y=4.88e6X+1.25e40.99905灭多威Y=1.70e6X+7.39e40.999110久效威Y=6.35e6X+2.95e40.998820涕灭威Y=7.04e6X+4.78e30.998515恶虫威Y=1.42e7X+1.02e40.996012残杀威Y=2.354e7X+2.40e50.99705克百威Y=9.81e6X+1.78e40.998810乙硫苯威Y=8.48e6X+1.22e40.99586异丙威Y=6.42e6X-9.0e3 0.99938仲丁威Y=5.12e6X+3.69e40.996712甲硫威Y=4.39e6X-4.59e30.998010猛杀威Y=8.80e6X-765 0.999510
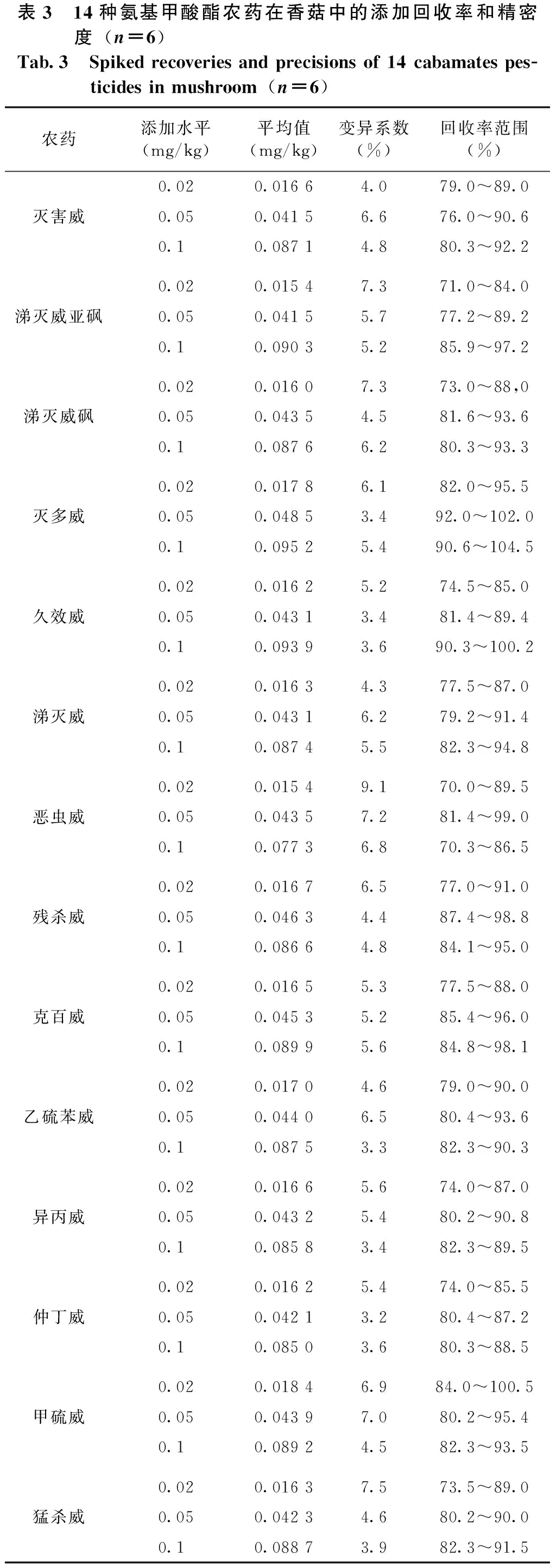
表3 14种氨基甲酸酯农药在香菇中的添加回收率和精密度(n=6)Tab.3 Spikedrecoveriesandprecisionsof14cabamatespes-ticidesinmushroom(n=6)农药添加水平(mg/kg)平均值(mg/kg)变异系数(%)回收率范围(%)灭害威0.020.01664.079.0~89.00.050.04156.676.0~90.60.10.08714.880.3~92.2涕灭威亚砜0.020.01547.371.0~84.00.050.04155.777.2~89.20.10.09035.285.9~97.2涕灭威砜0.020.01607.373.0~88,00.050.04354.581.6~93.60.10.08766.280.3~93.3灭多威0.020.01786.182.0~95.50.050.04853.492.0~102.00.10.09525.490.6~104.5久效威0.020.01625.274.5~85.00.050.04313.481.4~89.40.10.09393.690.3~100.2涕灭威0.020.01634.377.5~87.00.050.04316.279.2~91.40.10.08745.582.3~94.8恶虫威0.020.01549.170.0~89.50.050.04357.281.4~99.00.10.07736.870.3~86.5残杀威0.020.01676.577.0~91.00.050.04634.487.4~98.80.10.08664.884.1~95.0克百威0.020.01655.377.5~88.00.050.04535.285.4~96.00.10.08995.684.8~98.1乙硫苯威0.020.01704.679.0~90.00.050.04406.580.4~93.60.10.08753.382.3~90.3异丙威0.020.01665.674.0~87.00.050.04325.480.2~90.80.10.08583.482.3~89.5仲丁威0.020.01625.474.0~85.50.050.04213.280.4~87.20.10.08503.680.3~88.5甲硫威0.020.01846.984.0~100.50.050.04397.080.2~95.40.10.08924.582.3~93.5猛杀威0.020.01637.573.5~89.00.050.04234.680.2~90.00.10.08873.982.3~91.5
3 结论与讨论
本研究考察了以PSA和GCB为吸附剂的分散固相萃取法对食用林产品香菇中14种氨基甲酸酯类农药残留的提取的适用性,并建立了香菇中14种氨基甲酸酯类农药高效液相色谱-串联质谱的分析方法。实验结果表明,采用PSA和GCB作为分散固相萃取的混合吸附剂,能够有效地去除基质中的干扰成分,然后经HPLC-MS/MS在多反应监测模式下测定,外标法定量,14种目标物的平均加标回收率为70.0%~104.5%,相对标准偏差为3.2%~9.1%。采用分散固相萃取这种新型样品前处理技术比传统的农残分析方法更为快速和经济,具有很强的实用性,适用于食用林产品香菇中14种氨基甲酸酯类农药残留的检测。
[1] 文卫华,佘佳蓉.食用林产品质量安全问题与对策[J].湖南林业科技,2013,40(1):104-107.
[2] 俞秀玲,王佳,孙晓薇.我国食用林产品标准体系进展及存在问题[J].安徽农业科学,2012,40(1):229-230.
[3] 汪阳东.试论我国可食用林产品质量安全管理[J].国家林业局管理干部学院学报,2011(3):18-21,27.
[4] 刘世民,陈建宏,罗结.我国食品质量安全预警管理现状及应对研究[J].食品安全质量检测学报,2011(10):254-259.
[5] 沈庚晨.森林病虫害防治中的农药污染及对策[J].山西林业,2004(4):29-30.
[6] 邢礼国.正确使用农药防治森林害虫[J].辽宁林业科技,2004(4):19-30.
[7] 罗亮,岳永德,汤锋,等.重要食用林产品中农药多残留快速检测方法的研究[J].安徽农业大学学报,2011,38(1):72-80.
[8] 韩梅,侯雪.气相色谱串联质谱法测定香菇中几种氨基甲酸酯农药残留量[J].中国测试,2013,39(1):52-55.
[9] Mohammad S,Nafise E.Analysis of carbamate pesticides in water samples using single-drop microextraction and gas chromatography-mass spectrometry[J].Anal Bioanal.Chem.,2008,391:1091-1100.
[10] 丁晨红,骆冲,邓义才,等.分散固相萃取-反相高效液相色谱法测定蔬菜、水果中10种氨基甲酸酯类农药残留[J].热带农业科学,2014,34(4):77-82.
[11] Santalad A.,Srijaranai S,Burakham R,et al.Cloud-point extraction and reversed-phase-high-performance liquid chromatography for the determination of carbamate insecticide residues in fruits[J].Anal Bioanal.Chem.,2009,394(5):1307-1317.
[12] 姚家彪,赵颖,潘伟,等.蔬菜和食用菌中氨基甲酸酯类农药残留检测技术[J].应用化学,2010,27(4):488-493.
[13] 郭立,张清敏.氨基甲酸酯类农药残留分析方法的研究进展[J].天津农学院学报,2001,8(4):15-21.
[14] 陶传江.高效液相色谱柱后衍生法检测蔬菜中氨基甲酸酯类杀虫剂农药残留[J].农药科学与管理,2001,22(4):18-19.
[15] 赵亚华,何学芳,李勇,等.食品中40种有机磷和氨基甲酸酯农药多残留快速检测技术研究[J].中国卫生检验杂志,2007,17(11):1938-1940.
(文字编校:张 珉)
Determinationofcarbamatepesticideresiduesinedibleforestproducts
YANG Zhenxing1,2, DENG Jiehong1*, XI Hui2
(1.College of Food Science and Technology, Hunan Agricultural University, Changsha 410004, China;2.Changsha Environmental Protection College, Changsha 410004, China)
Developed a method for determination of 14 carbamate pesticides in edible forest product(mushroom)by dispersive solid-phase extraction cleanup and high performance liquid chromatography-tandem mass spectrometry.The sample was extracted by acetonitrile and purified by PSA powder with a little GCB powder.Then the target was determined and confirmed by HPLC-MS/MS using external standard method.The results indicated that the calibration curves of 14 carbamete pesticides showed good linear relationship in the concentration of 0.005~0.100 0 mg/L with correlation coefficients greater than 0.995 8.The recoveries of the 14 carbamate pesticides in mushroom(from 0.01 to 0.05 mg/kg )ranged from 70.0% to 104.5% with the relative standard deviation(RSD)ranged from 3.2% to 9.1%.The method was simple,fast and sensitive.It was also demonstrated that this analysis method was suitable for simultaneous determination of 14 carbamate pesticides in edible forest food.
carbamates; pesticide residues; forest food; high performance liquid chromatography-tandem mass spectrometry.
2014-11-10
湖南省教育厅优秀青年项目(14B085)。
杨振兴(1982-),男,黑龙江省牡丹江市人,硕士研究生,主要从事食品安全及其检测研究。
* 为通讯作者。
S 481.8
A
1003 — 5710(2015)01 — 0027 — 07
10. 3969/j. issn. 1003 — 5710. 2015. 01. 007
猜你喜欢
毛纺科技(2021年8期)2021-10-14
今日农业(2020年16期)2020-12-14
中国有色金属学报(2018年2期)2018-03-26
国际木业(2016年2期)2016-12-01
国际木业(2016年6期)2016-02-28
国际木业(2016年5期)2016-02-28
浙江林业(2015年1期)2015-11-30
中国塑料(2015年7期)2015-10-14
中华皮肤科杂志(2014年3期)2014-12-19
