
基于基因组序列的树干毕赤酵母生理特性解析
2015-11-11 04:01刘立明史仲平
生物加工过程 2015年1期
刘 婷,刘立明,史仲平
(1.江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122)
基于基因组序列的树干毕赤酵母生理特性解析
刘 婷1,2,刘立明1,史仲平2
(1.江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122;2.江南大学 工业生物技术教育部重点实验室,江苏 无锡 214122)
树干毕赤酵母作为一种潜在的纤维素乙醇生产菌株,有其独特的生理优势。然而与酿酒酵母生产乙醇相比,树干毕赤酵母生产纤维素乙醇还存在着诸多阻碍。为了从细胞整体水平进一步解析树干毕赤酵母的生理代谢特征,基于树干毕赤酵母的全基因组序列,利用生物信息学的分析算法,分别解析了树干毕赤酵母与酿酒酵母代谢网络的异同;利用流量平衡分析(FBA)验证了C源生长表型;预测了158个生长必需反应;鉴定了364个转录因子并对重要调控蛋白的功能进行了分析。
树干毕赤酵母;基因组序列;生理特性;流量平衡分析
传统的酿酒酵母(Saccharomyces cerevisiae)由于具有较高的乙醇产率、成熟的分子改造手段等一系列优点而成为乙醇工业化生产的最佳模式微生物[1]。乙醇生产的另一种优势菌株是运动发酵单胞菌(Zymomonas mobilis),其最高乙醇得率可达理论得率的97%,且对乙醇的耐受性高达120 g/L[2]。但对于纤维素乙醇的生产,上述2种微生物均存在的一个难以克服的缺陷,即不能天然发酵戊糖(半纤维素的主要成分)。尽管大量代谢工程改造工作以尝试导入木糖代谢途径,但是效果却不够理想[3]。
树干毕赤酵母 (Scheffersomyces stipitis),又名Pichia stipitis[4],是一种单倍体同宗配合的半子囊酵母,最早从腐木和寄生于林木的甲虫幼虫中分离得到[5]。树干毕赤酵母因其广泛的C源代谢能力而成为纤维素乙醇的潜在生产菌,在细胞中广泛存在的糖类分解酶系使得树干毕赤酵母能够代谢各种糖类分子。在所有从自然界中筛选出的木糖乙醇生产菌株中,树干毕赤酵母的代谢优势也十分显著:①木糖发酵能力强。单独发酵木糖生产乙醇的产率高达57 g/L[7],得率达0.47 g/g[8];②代谢副产物少。控制溶氧在一定水平内几乎检测不到木糖醇、甘油、乙酸等代谢物;③营养物质的需求简单。在无维生素的培养基中也能正常发酵;④对杂质的抵抗能力强;⑤能够代谢木质纤维素降解物中的毒性物质[6]。
与现今较为成熟的酿酒酵母乙醇发酵技术相比,树干毕赤酵母生产纤维素乙醇还存在着诸多阻碍:①与酿酒酵母的无氧发酵(crabtree-positive)生产乙醇的代谢方式不同,树干毕赤酵母在溶氧充足的条件下不能合成乙醇,只有控制溶氧在较低的水平(<1 mmol/(L·h))此菌株才能高效的生产乙醇[9],对溶氧精确的要求为工业化生产控制带来了难度;②树干毕赤酵母的底物发酵速率过低,以葡萄糖为例,酿酒酵母发酵速率达1.78 g/(g·h),而树干毕赤酵母葡萄糖发酵速率仅有0.39 g/(g·h)[10],木糖的发酵速率则更低,仅为0.23 g/(g·h)[11],过低的糖耗速率延长了发酵周期,提高了生产成本,不利于工业化生产;③与酿酒酵母相比,此菌株的遗传和生化背景不够清晰,代谢改造的成功率低。上述这些瓶颈问题需要一个系统水平的平台加以理解和解决,而微生物基因组规模代谢网络模型(genome-scale metabolic model,GSMM)提供了这样一个契机。
GSMM的一般研究思路是根据公布的测序结果进行基因组注释,建立详细的生化反应列表后转换为计算机可读的语言格式,再运用各种模拟算法对模型进行模拟预测和验证。自1999年流感嗜血杆菌的GSMM构建完成至今,相关研究工作广泛地在原核、真核微生物甚至人类基因组中开展,由此获得的数百个GSMM在生物、医药、食品等行业的基础研究方面产生良好效果[12]。基于约束的分析算法(constaint-based modeling,CBM)为GSMM提供了一个模拟平台,最大限度地了解微生物细胞的生理和代谢功能;通过流量平衡分析(FBA)、不同底物生长表型分析、基因敲除分析、流量可变分析、系统鲁棒性分析等不同的模拟算法,对细胞的表型进行量化预测[13]。笔者在前期研究中通过对树干毕赤酵母基因组注释并结合生物化学数据库和文献信息建立了以基因-蛋白质(酶)-生化反应关联为核心的基因组规模代谢网络模型iTL885[15]。该模型从糖类转运、C源代谢途径注释、溶氧角度阐述乙醇生成的影响因素,并提出了潜在的基因敲除靶点提高乙醇的生成速率。本文借助于树干毕赤酵母的基因组信息,利用生物信息学的分析算法,在细胞整体水平上进一步解析树干毕赤酵母的代谢特征。
1 材料与方法
1.1 基于COBRA工具箱的分析算法
从 In Silico Organisms 数 据 库 (http:// gcrg.ucsd.edu/InSilicoOrganisms/OtherOrganisms)中下载最近发表(截至2013年6月10日)的树干毕赤酵母基因组规模代谢网络模型iTL885。在Matlab平台上利用COBRA(constraint-based reconstruction and analysis)[16]进行模拟分析。作为目前应用最广泛的代谢网络模型分析手段,COBRA工具箱包含很多常用的分析算法。如流量平衡分析(flux balance analysis,FBA)主要用于预测细胞最大比生长速率和产物合成速率,而optimizeCbModel是COBRA工具箱中最常用的FBA算法。生长必需反应是指对细胞生长必需的、敲除则导致细胞不能存活的代谢反应,是一定生长条件下合成细胞组分所必需的反应集。生长必需反应的预测原理是运用singleRxnDeletion依次限制通过每个反应的流量为0,考察此时细胞最大比生长速率的变化。如果反应敲除后的比生长速率小于原菌的1.0×10-6,则被敲除的反应是必需反应,否则为非必需反应。
1.2 模拟条件选择与设定
对细胞生长表型的模拟需要预先设定模拟条件。模拟条件是实际生物学实验中细胞的培养环境。参考酿酒酵母等真核微生物的模拟条件,设置树干毕赤酵母模拟的基本培养基组分,包含基本营养元素、C源和O2。其中,对基本营养元素的供给没有限制,即对它们的交换反应的上下界分别设置为-1 000和1 000 mmol/(g·h),而对C源和O2则依据不同的培养条件需要限制它们所对应的不同的吸收速率。
1.3 转录因子(TF)的鉴定
①真菌转录因子数据库(Fungal Transcription Factors Database, FTFD, http://ftfd.snu.ac.kr/ index.php?a=view)运用标准的注释程序注释真菌基因组中的转录因子(transcription factors,TFs)[17],该数据库已收录171种真菌的66 392个假定转录因子(截至2013年6月10日)。在 FTFD中以Pichia stipitis为关键词检索并下载所有的转录因子,整理包括在FTFD数据中的名称(FTFD name)、所属TF家族名称(TF Family name)、基因座位名(Locus name)、序列长度(Length)和蛋白名称(Protein name)等信息。这些转录因子只是基于生物信息学算法的预测,准确性还有待验证。②在公布树干毕赤酵母测序结果的数据库(DOE Joint Genome Institute)树干毕赤酵母主页(http://genome.jgipsf.org/Picst3/Picst3.home.html)中根据GO分类,查找GO为0003700的transcription factor activity,获得139个比对的潜在转录因子。③为获取更加可靠的转录因子信息,运用本地BLAST对树干毕赤酵母全基因组进行比对(图1)。酿酒酵母专门的转录因子数 据 库 yeastract (http://www.yeastract.com/ index.php)收录了酿酒酵母112个经过验证的TFs及其调控的靶基因。下载该网站上所有酿酒酵母的转录因子,将该转录因子输入到Uniprot中通过ID Mapping将转录因子名称转化为Uniprot ID,再通过Retrieve下载相应fasta格式的蛋白质序列。将该序列与树干毕赤酵母的基因组蛋白质序列进行双向比对(BLASTp),比对结果的筛选条件设置为氨基酸序列相似度>35%和期望值(E值)<10-10。
图1 树干毕赤酵母转录因子预测的流程Fig.1 Flow chart of the Transcription Factors prediction in S.stipitis
2 结果与讨论
2.1 代谢网络特征分析
树干毕赤酵母基因组规模代谢网络模型iTL885由885个代谢基因、1 240个反应和870个代谢物构成[15](表1)。iTL885完整的内容已上传于In Silico Organisms数据库。由表1可知:选定酿酒酵母基因组规模代谢网络模型iMM904,比较2个模型注释出的基因总数目,发现iTL885的开放阅读框(ORFs)覆盖率是15.2%,高于iMM904(13.7%),证实了iTL885采用的整合基因组注释方法的全面性。由于酿酒酵母是一种典型的模式菌株,相关研究较为透彻,故iMM904包含的基因、反应、代谢物数目均大于iTL885(表1)。iTL885中约92%的代谢反应有基因关联,在760个有基因关联的反应中,118个代谢反应由多酶复合体构成的蛋白质催化,192个反应由同工酶催化,其余的450个反应由单个基因催化。不同于iMM904中的8个细胞分区,iTL885中的870个代谢物仅分布于细胞质(552个)、线粒体(148个)和细胞外(170个)3个区间,其中281个代谢物存在于不同的细胞区间。

表1 树干毕赤酵母和酿酒酵母模型比较Table 1 Model contents comparison of S.stipitis iTL885 and S.cerevisiae iMM904
2.2 树干毕赤酵母对C源的利用分析
通过FBA模拟了树干毕赤酵母在27种C源(15种糖类、9种醇类和3种酸)上的细胞生长表型。定性分析发现模拟结果和实际C源利用情况完全一致性(表2)。由表2可知:其中20种能够支持树干毕赤酵母生物量中所有前体物质的生成,证实了树干毕赤酵母广泛的C源代谢能力。有7种(棉子糖、山梨糖、蜜二糖、半乳糖醇、肌醇、甲醇和乳酸)不能支持生长,说明相关底物在树干毕赤酵母细胞中缺乏后续的代谢反应,致使生物量组分不能全部合成。上述模拟结果与数据中报道的树干毕赤酵母细胞在不同底物中的实际生长情况相一致。
选取3种典型C源(葡萄糖、木糖和鼠李糖),利用iTL885定量预测树干毕赤酵母的生长表型。由于缺乏准确的糖耗速率,在进行模拟时设定糖耗为相同的碳原子数。FBA预测的细胞生长和产物合成见表3。由表3可知:无论限制O2的吸收与否,细胞生长速率由大到小顺序为葡萄糖、木糖、鼠李糖,即葡萄糖为菌株生长最佳C源。当不限制O2的吸收速率时,树干毕赤酵母表现为完全的呼吸代谢,3种C源的代谢产物中除生物量和CO2外均没有其他代谢副产物,生物量生成速率快。当限制O2的吸收速率为4 mmol/(g·h)(以细胞干质量计)时,利用葡萄糖和木糖为C源可合成产物乙醇,且在模拟条件下木糖生成乙醇的速率(0.2548 mmol/(g·h))比葡萄糖(0.139 0 mmol/(g·h))更快,而鼠李糖则不能生成乙醇,是不可发酵C源,这与文献[19]的研究结果一致。

表2 树干毕赤酵母对不同C源底物的利用Table 2 Utilization of different substrates by S.stipitis
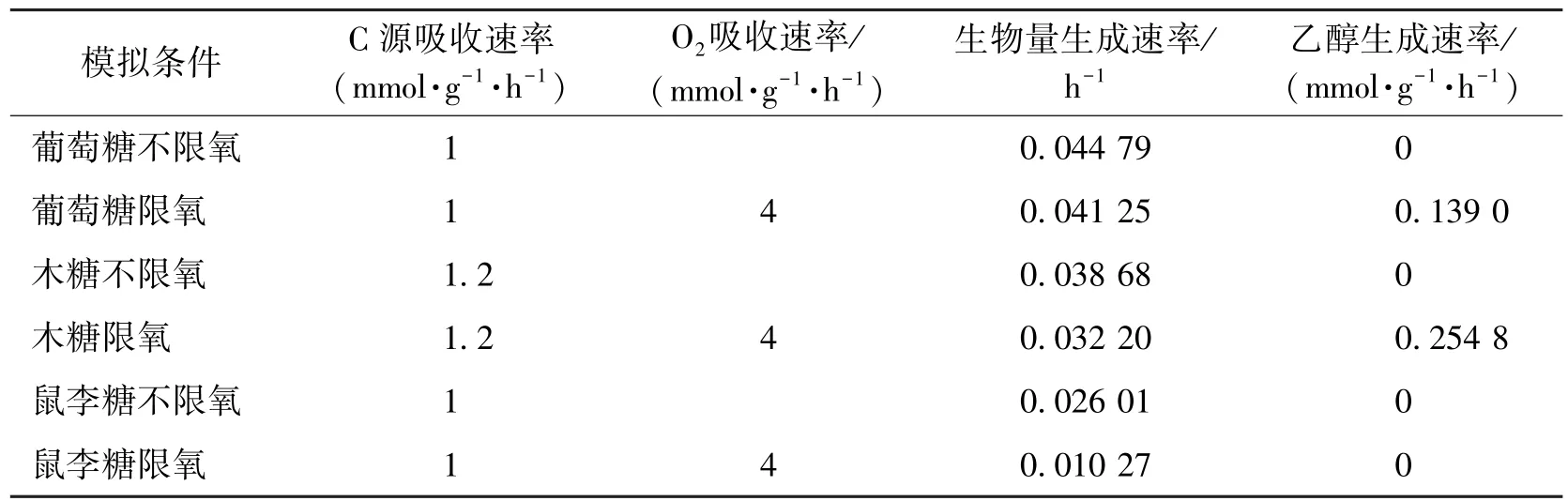
表3 在葡萄糖、木糖和鼠李糖3种培养基上细胞生长表型Table 3 Predicted growth phenotype of S.stipitis in glucose,xylose and rhamnose
2.3 树干毕赤酵母生长必需反应分析
基于iTL885预测在以葡萄糖为C源的基本培养基上树干毕赤酵母共有158个生长必需反应,占模型代谢反应总数的14.6%,此数值与报道的相同条件下酿酒酵母必需反应比例(13%)较接近[20]。树干毕赤酵母中,必需反应在6亚系统中分布情况见图2。由图2可知:氨基酸代谢(62个)、核苷酸代谢(22个)、脂质代谢(27个)、碳水化合物代谢(27个)、转运代谢(19个)辅因子和维生素代谢(1个)。其中氨基酸代谢占有的生长必需反应占必需反应总数的40%,它与C源核心代谢途径关联紧密,因而是最具刚性的代谢亚系统。树干毕赤酵母碳水化合物代谢所占生长必需反应的比例(14%)比酿酒酵母酿酒酵母比例(17%)小,表明2株菌在代谢特征上的差异,树干毕赤酵母比酿酒酵母拥有更广泛和活跃的代谢能力,存在较多相连通的支路途径,故对生长必需的反应数目较少。

图2 树干毕赤酵母(a)和酿酒酵母(b)在葡萄糖基本培养基上细胞生长必需反应分析Fig.2 Percentage of essential reactions in each subsystems on glucose minimal medium for(a)S.stipitis and(b)S.cerevisiae
2.4 转录因子的鉴定与功能分析
为获取最全面的树干毕赤酵母的转录因子信息、基于在线数据库和本地BLAST 3处数据来源对树干毕赤酵母的转录因子进行了预测(图3)。由图3可知:在FTFD中检索获得350个转录因子,约占基因组大小6%,分布在39个转录因子家族。同时在JGI树干毕赤酵母主页中根据GO信息查找获得139个转录因子。对二者的比较发现,GO获取的转录因子有6个不存在于FTFD中。运用BLASTp蛋白质序列双向比对,获得分布于16个转录因子家族的87个转录因子,比较发现有8个是FTFD未包括在内的。综合上述3种来源的转录因子,共计获得364个树干毕赤酵母转录因子,这为后续相关研究提供了备选转录因子库。不同来源获取的转录因子的可信度大小依次为BLAST、GO、FTFD。上述转录因子中还存在诸多功能未知的假定蛋白(putative proteins),因此对每个转录因子的准确功能和众多功能不明确的假定蛋白还需要生物学实验的验证。

图3 BLAST、GO、FTFD预测的转录因子及转录因子家族数目Fig.3 Number of transcription factors and transcription factors families obtained from BLAST,GO,and FTFD

图4 预测的转录因子数目及其家族分类Fig.4 Predicted transcription factors in some transcription factors families
上述方法获得的是所有可能存在于树干毕赤酵母中的转录因子,共分布于39个转录因子家族(TF Family),分布范围广。统计发现其中有6个转录因子家族包含的转录因子数目≥10个(图4)。由图4可知:锌簇转录因子(zinc finger)数目最多,共有10种类型构成的156个转录因子。排在其次的依次是OB折叠的核酸结合位点(nucleic acid-binding,OB-fold)转录因子家族(42个)、同源域(homeodomain-like,30个)、翼状螺旋的DNA结合结构域(winged helix repressor DNA-binding,20个)、Myb(15个)、bZIP(10个)这5个转录因子家族。
进一步对注释的树干毕赤酵母转录因子与酿酒酵母相关转录因子的功能进行比较,阐述在树干毕赤酵母中部分核心转录因子的代谢调控机制。按照序列同源相似性由高到低的顺序将不同转录因子及其调节功能列于表4。由表4可以发现:最具保守型的调控蛋白(氨基酸合成转录激活因子)GCN1、GCN4与酿酒酵母呈现一一对应的关系;树干毕赤酵母中与葡萄糖浓度感受相关的调节蛋白质有 RGT2、GRR1、SNF1、SKS1、SNF3、MIG2、MIG2.2和NRG1,这些调节蛋白与酿酒酵母中相对应的蛋白质表现出高度的序列结构相似性;同样具有高度保守性的有O2调节相关的蛋白质HAP5、HAP3和HAP2;不具有保守性或者保守性很低的蛋白质包括MIG1/CREA、ADR1、HAP1和HAP4。

表4 树干毕赤酵母与酿酒酵母同源的调控转录蛋白Table 4 Regulatory proteins in S.cerevisiae with corresponding proteins in S.stipitis
鉴于酿酒酵母与树干毕赤酵母代谢调节的差异,部分重要的调节蛋白只存在于酿酒酵母中,而在树干毕赤酵母中没有相对应的基因。这些酿酒酵母独有的调节基因包括INO2/INO4、RGT1、VID22和GCR1/GCR2。其中,INO2/INO4是磷脂代谢的转录激活因子,与乙醇耐受性相关[21];RGT1诱导葡萄糖转运蛋白(Hxt)表达;VID22参与糖酵解代谢途径中的果糖-1,6-二磷酸酶(FBP1)的转运和降解;而GCR1/GCR2则能够激活糖酵解相关基因的表达。较高的乙醇耐受性和糖酵解流量是酿酒酵母为适应高糖浓度发酵进化出来的机制。而树干毕赤酵母长期生存在低糖浓度环境中,其乙醇耐受性和糖酵解能力明显不足。
3 结论
基于作者前期构建的树干毕赤酵母基因组规模代谢网络模型iTL885从基因、反应、代谢物3个层次对树干毕赤酵母的代谢网络特征进行了分析。以iTL885为平台,运用FBA定性分析了树干毕赤酵母利用27种不同底物时细胞生长情况,与报道的细胞生长结果完全一致。选取木糖、葡萄糖和鼠李糖3种典型C源定量分析细胞生长和乙醇生成能力,发现细胞生长速率由大到小是葡萄糖、木糖、鼠李糖,且葡萄糖和木糖能发酵生成乙醇,鼠李糖是不可发酵C源。单反应敲除共预测到树干毕赤酵母有158个生长必需反应,与酿酒酵母在同样条件下的生长必需反应进行比较,证实了2株菌在代谢特征上的相同和差异之处。基于在线数据库和本地BLAST,获取分布于39个转录因子家族的364个树干毕赤酵母转录因子。通过与酿酒酵母相关转录因子的功能进行比较,证实酿酒酵母与树干毕赤酵母代谢调节的异同。
[1]Matsushika A,Inoue H,Kodaki T,et al.Ethanol production from xylose in engineered Saccharomyces cerevisiae strains:current state and perspectives[J].Appl Microbiol Biotechnol,2009,84(1):37-53.
[2]Seo J S,Chong H Y,Park H S,et al.The genome sequence of the ethanologenic bacterium ZymomonasmobilisZM4[J].Nat Biotechnol,2005,23(1):63-68.
[3]Kuhad R C,Gupta R,Khasa Y P,et al.Bioethanol production from pentose sugars:current status and future prospects[J]. Renew Sust Energ Rev,2011,15(9):4950-4962.
[4]Kurtzman C P,Suzuki M.Phylogenetic analysis of ascomycete yeasts that form coenzyme Q-9 and the proposal of the new genera Babjeviella, Meyerozyma, Millerozyma, Priceomyces, and Scheffersomyces[J].Mycoscience,2010,51(1):2-14.
[5]Toivola A,Yarrow D,van den Bosch E,etal.Alcoholic fermentation of D-xylose by yeasts[J].Appl Environ Microbiol,1984,47(6):1221-1223.
[6]Agbogbo F K,Coward-Kelly G.Cellulosic ethanol production using the naturally occurring xylose-fermenting yeast,Pichia stipitis[J]. Biotechnol Lett,2008,30(9):1515-1524.
[7]Ferrari M,Neirotti E,Albornoz C,et al.Ethanol production from eucalyptus wood hemicellulose hydrolysate by Pichia stipitis[J]. Biotechnol Bioeng,1992,40(7):753-759.
[8]Ligthelm M E,Prior B A,du Preez J C.The oxygen requirements of yeasts for the fermentation of D-xylose and D-glucose to ethanol[J].Appl Microbiol Biotechnol,1988,28(1):63-68.
[9]Grootjen D,van der Lans R,Luyben K.Effects of the aeration rate on the fermentation of glucose and xylose by Pichia stipitis CBS 5773[J].Enzyme Microb Tech,1990,12(1):20-23.
[10]Jeffries T W.Emerging technology for fermenting D-xylose[J]. Trends Biotechnol,1985,3(8):208-212.
[11]Slininger P,Bothast R,Okos M,et al.Comparative evaluation of ethanol production by xylose-fermenting yeasts presented high xylose concentrations[J].Biotechnol Lett,1985,7(6):431-436.
[12]Oberhardt M A,Palsson B Ø,Papin J A.Applications of genomescale metabolic reconstructions[J].Mol Syst Biol,2009,5(1). 320.doi:10.1038/msb.2009.77.
[13]刘立明,刘婷,邹伟.基因组规模代谢网络模型的约束算法及其应用[J].生物加工过程,2012,10(6):70-77.
[14]Feist A M,Palsson B Ø.The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli[J]. Nat Biotechnol,2008,26(6):659-667.
[15]Liu T,Zou W,Liu L,et al.A constraint-based model of Scheffersomyces stipitis for improved ethanol production[J]. Biotechnol Biofuels,2012,5(1):72.
[16]Becker S A,Feist A M,Mo M L,et al.Quantitative prediction of cellular metabolism with constraint-based models:the COBRA toolbox[J].Nat Protoc,2007,2(3):727-738.
[17]Park J,Jang S,Kim S,et al.FTFD:an informatics pipeline supporting phylogenomic analysis of fungal transcription factors[J].Bioinformatics,2008,24(7):1024-1025.
[18]Mo M L,Palsson B Ø,Herrgard M J.Connecting extracellular metabolomic measurements to intracellular flux states in yeast[J]. BMC Syst Biol,2009,3:37.
[19]Du Preez J,Bosch M,Prior B.The fermentation of hexose and pentose sugars by Candida shehatae and Pichia stipitis[J].Appl Microbiol Biotechnol,1986,23(3/4):228-233.
[20]Widiastuti H,Kim J Y,Selvarasu S,et al.Genome-scale modeling and in silico analysis of ethanologenic bacteria Zymomonas mobilis[J].Biotechnol Bioeng,2011,108(3):655-665.
[21]Ambroziak J,Henry S A.INO2 and INO4 gene products,positive regulators of phospholipid biosynthesis in Saccharomyces cerevisiae,form a complex that binds to the INO1 promoter[J].J Biol Chem,1994,269(21):15344-15349.
(责任编辑 荀志金)
Metabolic trait of Scheffersomyces stipitis based on genome sequence
LIU Ting1,2,LIU Liming1,SHI Zhongping2
(1.State Key Laboratory of Food Science and Technology,Jiangnan University,Wuxi 214122,China;2.The Key Laboratory of Industrial Biotechnology of the Ministry of Education,Jiangnan University,Wuxi 214122,China)
As a potential cellulosic ethanol producer,Scheffersomyces stipitis has its unique metabolic traits.However,problems need to be solved for Scheffersomyces stipitis cellulosic ethanol production compared with Saccharomyces cerevisiae ethanol production.Based on the genome data of Scheffersomyces stipitis,we did a further systematical analysis of the metabolic traits of Scheffersomyces stipitis was by bioinformatic algorithm.Genome-scale metabolic model(GSMM)similarity and difference between Scheffersomyces stipitis and Saccharomyces cerevisiae was compared.Cell growth phenotype on different carbon sources was validated with flux balance analysis.Furthermore,158 growth essential reactions were predicted on glucose minimal medium.Finally,364 transcription factors were identified and the function of important regulatory proteins were discussed.
Scheffersomyces stipitis;genome sequence;metabolic trait;flux balance analysis
Q78
A
1672-3678(2015)01-0075-07
10.3969/j.issn.1672-3678.2015.01.013
2013-06-09
教育部新世纪优秀人才支持计划(NCET-10-0456);江苏省杰出青年基金(BK2012002);中组部首批青年拔尖人才支持计划
刘 婷(1988—),女,江苏淮安人,硕士研究生,研究方向:工业微生物;史仲平(联系人),教授,E-mail:zpshi@jiangnan.edu.cn
猜你喜欢
酿酒科技(2021年8期)2021-12-06
军事文摘(2021年16期)2021-11-05
军事文摘·科学少年(2021年1期)2021-02-04
食品与发酵工业(2018年3期)2018-04-12
小猕猴学习画刊(2017年1期)2017-02-17
小猕猴学习画刊(2017年1期)2017-02-17
广东饲料(2016年1期)2016-12-01
工业微生物(2016年5期)2016-11-11
化学与生物工程(2016年10期)2016-11-10
故事作文·低年级(2016年7期)2016-05-14
