
基于高通量测序技术检测全基因组DNA甲基化水平的方法
2015-10-27 07:17马浪浪梁振娟先新罗仕文李清超梁黔云
生物技术通报 2015年7期
马浪浪梁振娟先新罗仕文李清超梁黔云
(1. 四川农业大学玉米研究所,成都 611130;2. 毕节市农业科学研究所,毕节 551700)
基于高通量测序技术检测全基因组DNA甲基化水平的方法
马浪浪1,2梁振娟2先新2罗仕文2李清超2梁黔云2
(1. 四川农业大学玉米研究所,成都 611130;2. 毕节市农业科学研究所,毕节 551700)
DNA甲基化的研究最近几年一直是表观遗传学研究的重点。有关DNA甲基化水平检测的方法大体上有十几种,随着第二代测序技术的发展,实现了在全基因组水平上对甲基化状态进行检测。目前,基于第二代高通量测序进行全基因组DNA甲基化水平检测的方法主要有BSP-seq(亚硫酸氢盐修饰结合直接测序法)、MeDIP-seq(甲基化DNA免疫共沉淀测序法)、MBD-seq(甲基化DNA富集结合高通量测序法)。就这3种方法在原理、流程、优缺点、优化使用及后期需要用到的部分生物信息学资源等方面的研究进展作一综述,旨在为研究者在采用高通量测序方法研究DNA甲基化模式时提供一些思路。
高通量测序技术;全基因组;DNA甲基化水平;BSP-seq;MeDIP-seq;MBD-seq
表观遗传学的研究日益深入,作为其中重要一方面的DNA甲基化更是长期研究的热点。人们越来越清楚地认识到有关DNA甲基化的研究很有可能在医学、农学等领域产生重大变革。
1 DNA甲基化的基本特征及维持机制
DNA甲基化是指S-腺苷甲硫氨酸上的甲基在甲基化转移酶的作用下转移到DNA胞嘧啶或腺嘌呤上,从而对DNA进行修饰,进而发生一系列的表观修饰现象[1]。
与其他研究相似,人们对于植物DNA甲基化修饰的研究始于模式植物拟南芥,拟南芥全基因组甲基化图谱(单核苷酸分辨率)已经公布[2-4]。研究表明,在植物中发生DNA甲基化修饰的区域主要有CpG岛、CHH、CHG(H代表A或T)以及转录区域、基因主体上[2-7]。发生于CpG岛、CHH、CHG区域的DNA甲基化修饰主要生物学功能为:通过产生甲基化重复序列来抑制基因组DNA,从而达到保护基因组的目的;发生于转录区域、基因主体区域的DNA甲基化修饰主要生物学功能为:通过甲基化水平的高低调节基因转录水平。一系列研究都表明,基因的甲基化水平与转录水平呈负相关性[4,6,8]。
DNA甲基化能引起一系列表观遗传修饰现象:植物开花、衰老、抗逆、基因沉默及杂种优势等。如Sheldon等[9]研究表明抽薹相关基因的甲基化水平降低,可以造成植物提取开花。Fraga等[10]研究发现成年辐射松植株的甲基化程度明显高于幼年植株的甲基化程度。Roberta等[11]对三叶草根系、大麻根系进行重金属处理后发现它们的基因组DNA甲基化水平均降低。Peerbolte等[12]研究表明在T-DNA上的冠瘿碱基因发生甲基化作用,使得该基因在随后的转化过程中沉默。孙其信[13]对植物分别进行自交、杂交,结果表明自交后基因组DNA甲基化水平增加,杂交后基因组DNA甲基化水平降低。本课题组以玉米幼胚胚性愈伤组织诱导过程为切入点,对该过程中不同时期全基因组DNA甲基化模式进行了分析(运用了甲基化DNA免疫共沉淀结合高通量测序技术,即MeDIP-seq,该技术将会在后文阐述),得出了在这一过程中DNA甲基化水平的变化情况,具体研究结果待发表。以上几个例子表明:在一系列的生物学过程中都有DNA甲基化的参与,随着研究方法、技术的提高,研究者对于DNA甲基化参与表观遗传修饰的研究必将更加深入。
目前,有关DNA甲基化的维持机制主要基于4类甲基转移酶进行作用。如MET1甲基转移酶在DNA复制过程中能识别单拷贝及重复序列的CG位点,使得在该CG位点发生甲基化作用[14,15];DRM重新甲基化酶则是在小分子RNA介导下识别胞嘧啶序列,进而发生重新甲基化作用,即RdDM[16,17];CMT染色质甲基化酶可以特异性作用于异染色质区的CNG序列,进而发生甲基化作用,该酶在植物中特有存在[18-20];研究者认为DNMT2家族的一些同系物可能也参与甲基化的维持机制,具体方式有待进一步研究[21]。这几类甲基转移酶有时也并非独立参与机制的调控,它们常常相互作用,共同参与机制的调控。我们相信,随着研究技术的不断提升,研究者对于DNA甲基化的维持机制将会有更加全面的认识。
2 基于全基因组进行DNA甲基化检测的方法
目前,研究者对于DNA甲基化的检测方法主要可以分为两个层面:(1)基于全基因组DNA甲基化水平检测;(2)基于片段(位点)DNA甲基化检测。基于全基因组DNA甲基化的检测方法,见表1。
上述基于基因组DNA甲基化的研究方法中,BSP-seq、MeDIP-seq和MBD-seq这3种方法随着全基因组高通量测序技术的发展以及生物信息学的兴起,在人们的研究中越来越受到重视。故下文主要针对这3种方法进行介绍。
2.1 三种检测方法的工作流程
2.1.1 BSP-seq法检测DNA甲基化水平 BSP-seq法最早是由Frommer等[28]提出,其检测DNA甲基化的原理已在表1中介绍。该方法的检测流程如下:(1)基因组DNA的提取、检测、纯化(保证DNA的质量、纯度等);(2)用亚硫酸氢钠处理基因组DNA;(3)对修饰后的DNA进行回收并纯化;(4)引物设计(根据目的基因启动子序列);(5)PCR扩增目的片段;(6)PCR产物克隆、测序;(7)对所得序列进行比对、对所得数据进行生物信息学分析[28,31]。
运用BSP-seq检测DNA甲基化需要注意的问题:(1)样品的质量应适中,DNA量太多会造成亚硫酸盐修饰不完全,DNA量太少则会造成后期实验回收量不足;(2)在亚硫酸盐修饰过程中必须保证pH值绝对准确、且需要多次摸索合适的反应时间;(3)因为该过程涉及到PCR扩增,故引物设计、PCR反应体系都需不断优化[32]。此外,在后期数据处理时可能会碰到一些问题,如由于测序错误、污染等情况,得到的数据处理时比较麻烦。针对这些情况,Krueger等[33]给出一些处理方法,使得实验结果的准确性提高。
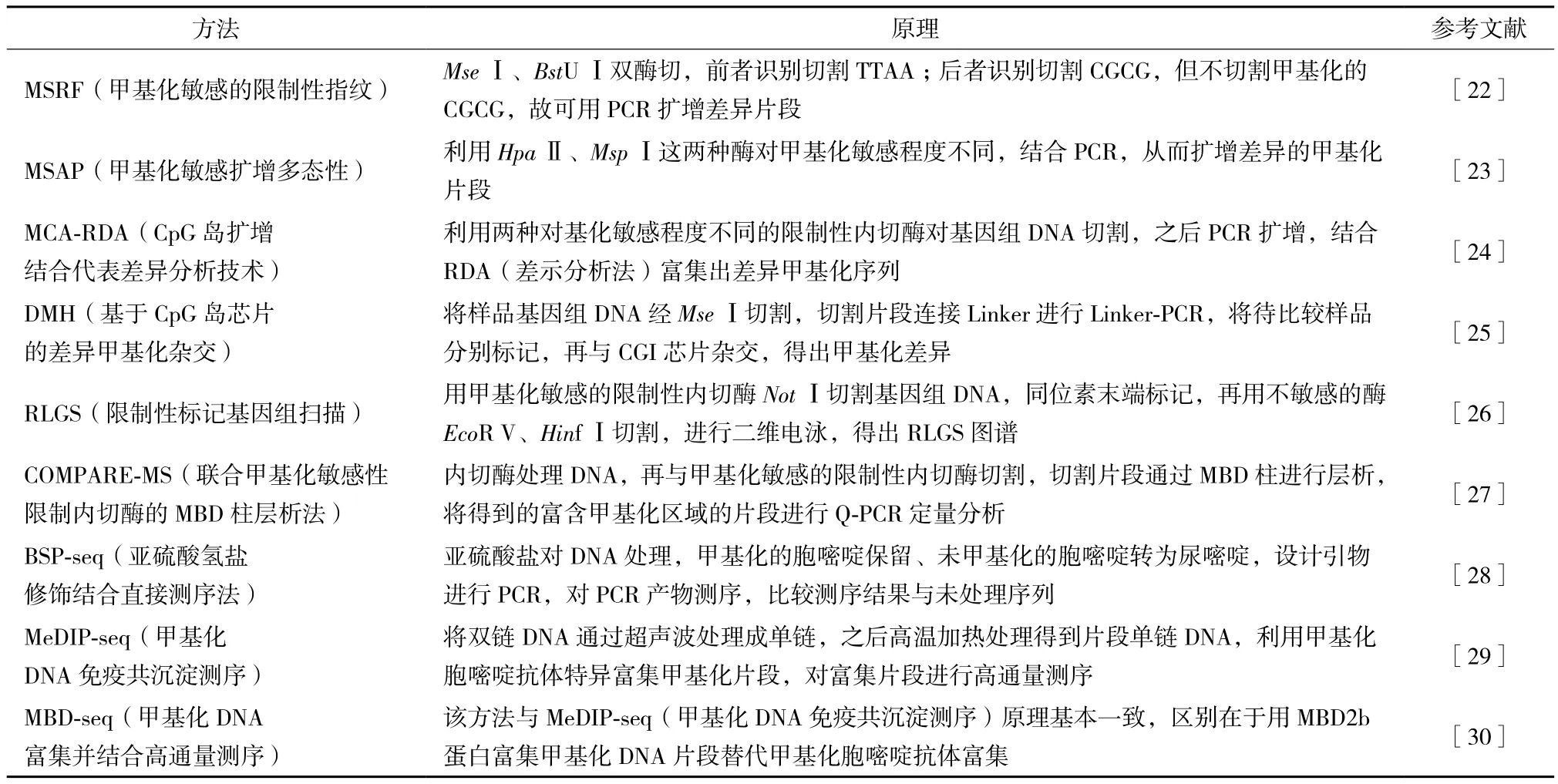
表1 基于全基因组进行DNA甲基化的检测方法
2.1.2 MeDIP-seq法检测DNA甲基化水平 MeDIP-seq法是由Weber等[34]在2005年首先提出,是指5'-甲基胞嘧啶抗体特异富集基因组DNA上发生甲基化的片段,之后通过对这些富集的甲基化片段进行高通量测序,从而获得基因组DNA的甲基化发生区域、甲基化水平等。具体操作流程为:(1)通过超声波将DNA打断成DNA片段;(2)通过高温使得片段DNA解为单链;(3)片段DNA与5'-甲基胞嘧啶抗体特异反应,从而构建甲基化DNA文库(这一过程伴随着对片段DNA进行PCR等处理);(4)高通量测序及后期对数据进行生物信息学分析(测序数据去污染、去接头、统计数据产出量;测序序列与参考基因组序列比较;测序数据在基因组的分布;测序数据富集区(Peak区)的信息统计;基于Peak进行全基因组甲基化差异分析等)[29,34]。运用该方法进行基因组DNA甲基化检测需要注意:(1)DNA片段富集过程中富集条件必须摸索才能达到好的效果;(2)DNA片段富集后,当富集量较少时需采用PCR扩增,PCR反应体系需不断优化;(3)要保证样品在整个测序过程中无污染等[35]。
2.1.3 MBD-seq法检测DNA甲基化水平 MBD-seq法检测基因组DNA甲基化水平的原理与MeDIP-seq法基本相似。其检测流程如下:(1)特异性结合甲基化DNA的蛋白MBD2b富集高甲基化的DNA片段;(2)对富集的DNA片段进行末端补平得到平末端片段;(3)对平末端片段进行末端加A',得到3'-dA黏性末端;(4)加连接头,获得带接头片段,进而得到长度合适的目的DNA片段;(5)进行PCR扩增,构建MBD-seqDNA文库;(6)进行高通量测序,并对所得结果进行生物信息学分析[30]。
近年,研究者采用该方法对DNA甲基化模式进行了大量的研究,如Wang等[36]采用MBD-seq法分析家族腺瘤性息肉症患者在BRB栓剂的治疗下的甲基化状态显示,在该栓剂的作用下出现较多的脱甲基转录位点;家族腺瘤性息肉症患者的息肉减少。综合该结果表明,家族腺瘤性息肉症可能与脱甲基化转录相关。运用该方法进行基因组DNA甲基化水平检测时,需要注意的问题同运用MeDIP-seq法需要注意的问题基本相似。
2.2 三种检测方法的比较及优化使用
BSP-seq、MeDIP-seq、MBD-seq这3种基于高通量测序检测全基因组DNA甲基化方法在费用、分辨率、数据分析等方面有一定的差异。表2是就BSP-seq、MeDIP-seq和MBD-seq这3种方法在这些方面的差异进行的总结。
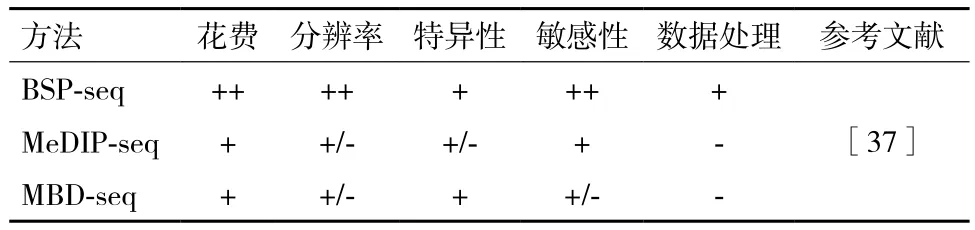
表2 三种基于高通量测序检测全基因组DNA甲基化方法比较
由于BSP-seq法既可得到全基因组上的甲基化信息,又可得到特异位点上的甲基化信息,故此法被认为是金标准。但对于基因组较大的物种而言,该方法花费巨大,且高通量测序数据也很庞大,对于后期数据处理、分析提出了挑战。因此,研究者在实际操作中往往很难接受或采用该方法。MeDIP-seq法及MBD-seq法测序费用相对较低,数据量较小,但分析得到的结果很难验证。基于上述原因,科研工作者进行了大量的改进尝试。如Li等[38]采用BSP-seq法得出人类外周血单核细胞中的一个单碱基分辨率甲基化组,并以此图谱作为参考基因组对比了MeDIP-seq法及MBD-seq法的实验结果,结果显示,MeDIP-seq对高度甲基化、高CpG密度更加敏感;MDB-seq对高度甲基化、中等CpG密度更加敏感。因此,这两种方法可以形成很好的互补效应。
2.3 三种检测方法所涉及的生物信息学分析资源BSP-seq、MeDIP-seq、MBD-seq这3种方法在经过高通量测序后得到大量的数据。对于这些数据需要进行进一步的生物信息学分析,才能得到研究者理想的结果,详见表3。

表3 部分生物信息学分析资源
3 展望
随着第二代高通量测序的发展,人们将在越来越多的领域中使用它。DNA甲基化作为表观遗传学中重要的一个方面,在近几年的研究中借助第二代高通量测序技术取得了长足的进展。采用高通量测序技术研究DNA甲基化模式将是一种趋势。
BSP-seq、MeDIP-seq、MBD-seq是目前基于高通量测序技术研究全基因组DNA甲基化模式的3种方法。正如前文所述,这3种方法各有优劣,结合花费、分辨率、特异性、敏感度及数据分析等方面研究者认为[40]:在针对一些基因组较大的物种进行全基因组DNA甲基化模式研究中采用MeDIP-seq法和MBD-seq法相互补充较优于采用BSP-seq法;而在针对一些基因组较小的物种进行研究时,可以根据研究目标选择不同的方法(如需要得到高分辨率、高特异性及高敏感度等较高要求时可优先选择BSP-seq法,反之可选择MeDIP-seq法或MBD-seq法)。
本课题组采用MeDIP-seq法研究了高胚诱导率玉米骨干自交系幼胚发育过程中全基因组DNA甲基化模式,结合近几年拟南芥全基因组DNA甲基化已公布图谱,笔者以为,应以此为模式并结合优化使用BSP-seq、MeDIP-seq、MBD-seq这3种方法开展相关植物全基因组DNA甲基化研究,相信在不久的将来有关DNA甲基化的确切机制将会被人类解析。
[1] Bird AP. CpG-rich islands and the function of DNA methylation[J]. Nature, 1985, 321(6067):209-213.
[2]Cokus SJ, Feng S, Zhang X, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning[J]. Nature, 2008, 452(7184):215-219.
[3]Lister R, O’Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis[J]. Cell, 2008, 133(3):523-536.
[4]Zhang X, Yazaki J, Sundaresan A, et al. Genome-wide highresolution mapping and functional analysis of dna methylation in Arabidopsis[J]. Cell, 2006, 126(6):1189-1201.
[5]Tran RK, Henikoff JG, Zilberman D, et al. DNA Methylation profiling identifies CG methylation clusters in Arabidopsis genes[J]. Current Biology, 2005, 15(2):154-159.
[6]Zilberman D, Gehring M, Tran RK, et al. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription[J]. Nature Genetics, 2006,39(1):61-69.
[7]Hsieh TF, Ibarra CA, Silva P, et al. Genome-wide demethylation of Arabidopsis endosperm[J]. Science, 2009, 324:1451-1454.
[8]Zemach A, McDaniel IE, Silva P, et al. Genome-wide evolutionary analysis of eukaryotic DNA methylation[J]. Science, 2010, 328(5980):916-919.
[9] Sheldon CC, Finnegan EJ, Rouse DT, et al. The control of flowering by vernalization[J]. Curr Opin Plant Biol, 2000, 3(5):418-422.
[10]Fraga MF, Cañal M, Rodríguez R. Phase-change related epigenetic and physiological changes in Pinus radiata D. Don[J]. Planta,2002, 215(4):672-678.
[11]Aina R, Sgorbati S, Santagostino A, et al. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp[J]. Physiologia Plantarum, 2004, 121(3):472-480.
[12]Peerbolte R, Leenhouts K, Hooykaas-van Slogteren GM, et al. Clones from a shooty tobacco crown gall tumor II:irregular T-DNA structures and organization, T-DNA methylation and conditional expression of opine genes[J]. Plant Mol Biol, 1986, 7:285-299.
[13]孙其信. 农作物杂种优势机理研究及展望[J]. 作物杂志,1998, 4:31.
[14] Bernstein BE, Meissner A, Lander ES. The mammalian epigenome[J]. Cell, 2007, 128(4):669-681.
[15] Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases[J]. Annu Rev Biochem, 2005, 74:481-514.
[16] Chan SW, Zilberman D, Xie Z, et al. RNA silencing genes control de novo DNA methylation[J]. Science, 2004, 303(5662):1336-1336.
[17]Cao X, Aufsatz W, Zilberman D, et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation[J]. Current Biology, 2003, 13(24):2212-2217.
[18] Papa CM, Springer NM, Muszynski MG, et al. Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation[J]. The Plant Cell Online, 2001, 13(8):1919-1928.
[19] Bartee L, Malagnac F, Bender J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene[J]. Genes & Development, 2001, 15(14):1753-1758.
[20] Lindroth AM, Cao X, Jackson JP, et al. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation[J]. Science, 2001, 292(5524):2077-2080.
[21]Goodrich J, Tweedie S. Remembrance of things past:chromatin remodeling in plant development[J]. Annual Review of Cell and Developmental Biology, 2002, 18(1):707-746.
[22]Huang THM, Laux DE, Hamlin BC, et al. Identification of DNA methylation markers for human breast carcinomas using the methylation-sensitive restriction fingerprinting technique[J]. Cancer Research, 1997, 57(6):1030-1034.
[23]Reyna-Lopez GE, Simpson J, Ruiz-Herrera J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms[J]. Molecular and General Genetics MGG, 1997, 253(6):703-710.
[24]Toyota M, Ho C, Ahuja N, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification[J]. Cancer Res, 1999, 59:2307-2312.
[25]朱燕. DNA 甲基化的分析与状态检测[J]. 现代预防医学,2006, 32(9):1070-1073.
[26] Hatada I, Hayashizaki Y, Hirotsune S, et al. A genomic scanningmethod for higher organisms using restriction sites as landmarks[J]. Proc Natl Acad Sci USA, 1991, 88(21):9523-9527.
[27] Yegnasubramanian S, Lin X, Haffner MC, et al. Combination of methylated-DNA precipitation and methylation-sensitive restriction enzymes(COMPARE-MS)for the rapid, sensitive and quantitative detection of DNA methylation[J]. Nucleic Acids Research, 2006, 34(3):e19.
[28] Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands[J]. Proc Natl Acad Sci USA, 1992,89:1827-1831.
[29] Thu KL, Vucic EA, Kennett JY, et al. Methylated DNA immunoprecipitation[J]. J Vis Exp:JoVE, 2009(23):935.
[30] Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome[J]. Nucleic Acids Research,2010, 38(2):391-399.
[31] Beck S, Rakyan VK. The methylome:approaches for global DNA methylation profiling[J]. Trends Genet, 2008, 24(5):231-237.
[32]http://www. biovol. net/mutation/12852. htm. 上海拜沃生物科技有限公司分子生物学技术服务, 2013-1-29
[33]Krueger F, Kreck B, Franke A, et al. DNA methylome analysis using short bisulfite sequencing data[J]. Nature Methods, 2012,9(2):145-151.
[34]Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells[J]. Nature Genetics, 2005, 37(8):853-862.
[35]http://www. plob. org/2012/01/09/1614. html.
[36]Wang LS, Burke C, Hasson H, et al. A phase 1b study of the effects of black raspberries on rectal polyps in patients with familial adenomatous polyposis[J]. Cancer Prev Res, 2014, 7:666-674.
[37] Mensaert K, Denil S, Trooskens G, et al. Next-generation technologies and data analytical approaches for epigenomics[J]. Environmental and Molecular Mutagenesis, 2014, 55(3):155-170.
[38] Li N, Ye M, Li Y, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology[J]. Methods,2010, 52(3):203-212.
[39] Rohde C, Zhang Y, Jurkowski TP, et al. Bisulfite sequencing data presentation and compilation(BDPC)web server—a useful tool for DNA methylation analysis[J]. Nucleic Acids Research, 2008,36(5):e34.
[40] Xi Y, Li W. BSMAP:whole genome bisulfite sequence MAPping program[J]. BMC Bioinformatics, 2009, 10(1):232.
[41] Down TA, Rakyan VK, Turner DJ, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis[J]. Nature Biotechnology, 2008, 26(7):779-785.
[42] Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers[J]. Bioinformatics, 2004,20(7):1170-1177.
[43]Takai D, Jones PA. The CpG island searcher:a new WWW resource[J]. In Silico Biology, 2003, 3(3):235-240.
[44]Ioshikhes IP, Zhang MQ. Large-scale human promoter mapping using CpG islands[J]. Nature Genetics, 2000, 26(1):61-63.
[45]Grunau C, Renault E, Rosenthal A, et al. MethDB-a public database for DNA methylation data[J]. Nucleic Acids Research, 2001, 29(1):270-274.
[46]Grunau C, Renault E, Roizes G. DNA methylation database“MethDB”:a user guide[J]. J Nutr, 2002, 132:2435S-2439S.
[47]Amoreira C, Hindermann W, Grunau C. An improved version of the DNA Methylation database(MethDB)[J]. Nucleic Acids Research, 2003, 31(1):75-77.
[48] Negre V, Grunau C. The MethDB DAS server:adding an epigenetic information layer to the human genome[J]. Epigenetics, 2006, 1(2):101-105.
[49]Kumaki Y, Oda M, Okano M. QUMA:quantification tool for methylation analysis[J]. Nucleic Acids Research, 2008, 36(suppl 2):W170-W175.
[50] Pattyn F, Hoebeeck J, Robbrecht P, et al. methBLAST and meth-PrimerDB:web-tools for PCR based methylation analysis[J]. BMC Bioinformatics, 2006, 7(1):496.
[51]Li LC, Dahiya R. MethPrimer:designing primers for methylation PCRs[J]. Bioinformatics, 2002, 18(11):1427-1431.
[52]Laird PW. Principles and challenges of genome-wide DNA methylation analysis[J]. Nature Reviews Genetics, 2010, 11(3):191-203.
(责任编辑 狄艳红)
Detection Methods of Genome-wide DNA Methylation Level Based on High-throughput Sequencing Technology
Ma Langlang1,2Liang Zhenjuan2Xian Xin2Luo Shiwen2Li Qingchao2Liang Qianyun2
(1. Maize Research Institute of Sichuan Agricultural University,Chengdu 611130;2. Bijie Institude of Agricultural Science,Bijie 551700)
DNA methylation has been being the research focus of epigenetics study in the recent years. There are over 10 kinds of methods for detecting DNA methylation level, and detection of methylation status on genome-wide level has been achieved with the development of the NGS(Next-Generation Sequencing). At present the major detection methods of DNA methylation level on genome-wide scope based on NGS include BSP-seq(Bisulfite Sequencing PCR), MeDIP-seq(Methylated DNA Immunoprecipitation Sequencing)and MBD-seq(Methylated DNA Binding Domain Sequencing). In this paper, the research progress of principles, procedures, advantages and disadvantages, optimization of the usage and some bioinformatics resources on these 3 methods were reviewed, which aims to provide some beneficial ideas to researchers who are studying DNA methylation models while employing methods of high-throughput sequencing.
high-throughput sequencing technology;genome-wide;DNA methylation level;BSP-seq;MeDIP-seq;MBD-seq
10.13560/j.cnki.biotech.bull.1985.2015.07.007
2014-10-19
贵州省科学技术基金计划重点项目[黔科合JZ字(2014)2001号],州省科学技术基金计划项目[黔科合J字(2013)2002号],贵州省毕节市农业攻关项目(毕科合19号)
马浪浪,男,硕士,研究方向:表观遗传学;E-mail:sxyljxml@163.com
李清超,男,助理研究员,研究方向:玉米分子育种;E-mail:569733223@qq.com梁黔云,男,研究员,研究方向:玉米育种;E-mail:blqy678@163.com
猜你喜欢
国际太空(2023年1期)2023-02-27
现代畜牧科技(2021年4期)2021-07-21
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
中国博物馆(2018年2期)2018-12-05
中国发展观察(2017年8期)2017-04-26
现代检验医学杂志(2015年4期)2015-02-06
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
遗传(2014年3期)2014-02-28
