
新型高能量密度化合物BNFDAONAB的结构与性能量子化学研究
2015-10-22 03:56荆苏明刘玉存刘登程郭嘉昒
火工品 2015年2期
荆苏明,刘玉存,刘登程,郭嘉昒
(中北大学化工与环境学院,山西太原,030051)
新型高能量密度化合物BNFDAONAB的结构与性能量子化学研究
荆苏明,刘玉存,刘登程,郭嘉昒
(中北大学化工与环境学院,山西太原,030051)
设计了一种新型高能量密度化合物(HEDC)—N,N、-二(4-硝基呋咱基-3-基-)-4,4´-二氨基-2、2´,3、3´,5、5´,6、6´-八硝基偶氮苯(BNFDAONAB),采用密度泛函理论(DFT)方法,在B3LYP/6-31+G**基组水平下对其结构进行优化并计算获得了其红外(IR)光谱;采用Monte-Carlo方法预测了BNFDAONAB的理论密度为2.08g/cm3;设计等键反应计算了生成焓为865.79kJ/mol;运用Klmet-Jacobs公式预测了BNFDAONAB的爆速、爆压和爆热值分别为9.13km/s、39.03GPa和4 487.44J/g;运用Keshavarz公式预测撞击感度H50为1.06cm;并利用逆合成分析法设计了其合成路线。结果表明,该化合物主要性能的预测值基本达到了HEDC的要求,是一种潜在的绿色起爆药。
起爆药;绿色;BNFDAONAB;密度泛函理论;爆轰性能
现代军事科学技术和高科技装备的发展,对其配套的弹药技术有了更高的要求,在此背景下,高能量密度化合物(HEDC)成为全世界含能材料领域密切关注的焦点之一[1-2]。各种HEDC不断出现,如早期的HMX,1,3,4,6-四硝基甘脲(TNGU)[3],2,5,7,9-四硝基-2,5,7,9-四氮杂双环[4.3.0]辛烷-8-酮,2,4,6,8-四氢-2,4,6,8-四氮杂双环[3.3.0]辛烷-3-酮,到现在的3,3’-二硝基氧化偶氮呋咱(DNOAF)[4]、六硝基六氮杂异伍兹烷(CL-20)和八硝基立方烷(ONC)等。这些化合物虽然在爆轰性能上表现优异,但都存在各种各样的缺陷,如合成方法繁冗复杂(ONC)、合成成本高(HMX、CL-20)、水稳定性差(TNGU)等。本文在保持化合物高能量、高密度的性能基础上,以高稳定性、简易合成条件和原料来源广泛为目的,基于偶氮联苯为基本单元设计了一种国内外未见报道的含能化合物—N,N-二(4-硝基呋咱基-3 -)-4,4´-二氨基-2、2´,3、3´,5、5´,6、6´-八硝基偶氮苯,并通过量子化学方法预测了其结构和性质。
1 计算方法与原理
相对于传统的从头算方法及半经验计算方法,B3LYP在保持了从头算的诸多优点的同时还考虑了电子相关,对分子性质的相关描述优于自洽场从头算,可在高精度基组水平上获得和实验值较为接近的分子结构和性能,且所需的计算量较小,因此在含能材料领域应用广泛[5-7]。本文运用gaussian 09程序包,采用密度泛函理论(DFT)的B3LYP方法[3],在6-31+G**基组水平上对N,N、-二(4-硝基呋咱基-3-基-)-4,4´-二氨基-2、2´,3、3´,5、5´,6、6´-八硝基偶氮苯(BNFDAONAB)进行几何结构全优化,如图1所示。
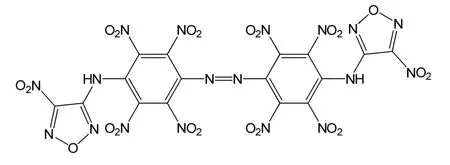
图1 BNFDAONAB化学结构图Fig.1 Chemical structure of BNFDAONAB
获得其稳定的构型和对应的红外(IR)光谱,在此优化结构基础上计算得到其自然轨道原子电荷以及前线轨道能量;采用Monte-Carlo[8]方法计算体积,进一步得到理论密度;设计等键反应[9-10]计算生成热;基于理论密度以及生成焓,运用Kamlet-Jacobs[11]公式预测了爆速、爆压和爆热等爆轰性能;计算过程中所有优化结构无虚频,均为势能面上的真实能量最小点。以上计算中所有收敛精度均取程序设定的内定值。
2 结果与讨论
2.1几何构型
BNFDAONAB优化后的构型及原子编号见图2,振动分析表明,优化后构型无虚频,表明该构型为势能面上的极小点,是相对稳定的结构。由图2可见,与偶氮苯直接相联的所有原子都处于同一平面,由于硝基的空间位阻效应,导致所有硝基和呋咱都发生不同程度的扭曲,这在一定程度上减小了分子内部集团间的相互排斥作用,有利于分子结构的稳定。

图2 优化后BNFDAONAB分子结构图Fig.2 Geometricconfiguration ofBNFDAONABafter optimization
另一方面,C-N和N-N单键的标准键长分别为0.147 0nm和0.140 0nm,硝基中的N-O键键长为0.120 0nm,表1为BNFDAONAB分子的部分键长。通过表1发现:偶氮苯环上的C-N和N-N键长为0.128 7~0.144 0nm,均介于正常双键(0.122nm)和单键(0.147nm)之间,趋于平均化;而与苯环直接相连的基团中,C-N单键的键长都有所收缩,键长变短,表明偶氮联苯环与其取代基中心原子联接紧密,结构紧凑,环稳定性较高,而N-O键普遍拉长,呋咱环中N-O键拉长的尤为明显,易于断裂,可能为热键引发键。
2.2振动与红外光谱
BNFDAONAB红外光谱的频率计算和强度的计算结果见图3(矫正系数为0.96)。由图3可见,该化合物主要有以下特征吸收峰:氢原子质量最小,3 000 cm-1左右的强吸收峰为C-H键对称和不对称收缩振动;1 598 cm-1处的强吸收峰为苯环的对称收缩振动所引起,1 536 cm-1处的强吸收峰为苯环的不对称收缩振动所引起,指纹区内1 120~1 170 cm-1处的中强吸收峰为硝基的中N=O的收缩振动,230~1 100 cm-1处的弱吸收峰主要对应C-H、N-H和C-NO2等键的完全振动,以及苯和呋咱环骨架的变形振动。
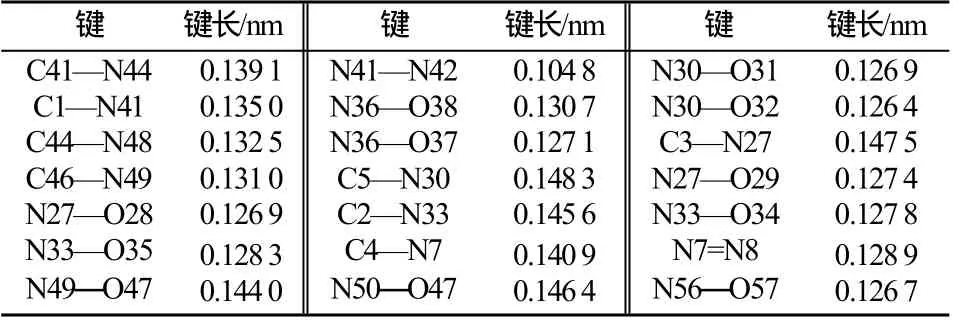
表1 BNFDAONAB分子部分键长Tab.1 Calculated bond length of BNFDAONAB
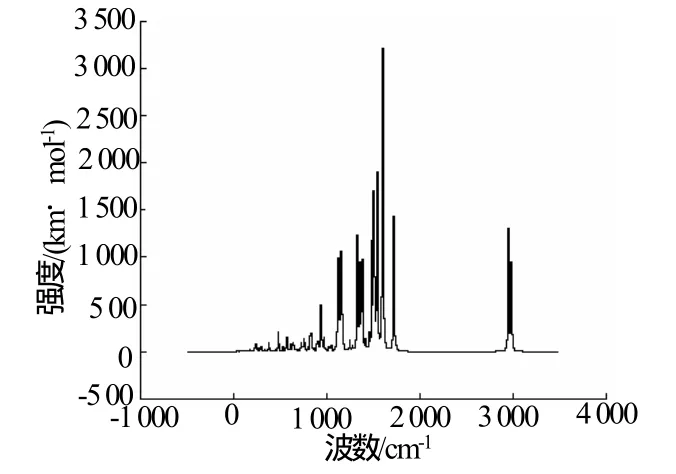
图3 BNFDAONAB分子红外光谱图Fig.3 Calculated IR spectrum of BNFDAONAB
该化合物为国内外尚未合成的新化合物,故无实验数据比较,但一些计算实践[12-13]证明,在DFT/ B3LYP下获得的红外数据非常可靠。
2.3分子前线轨道
在B3LYP/6-31+G**基组水平下,通过guassian09计算获得了BNFDAONAB的最高占有轨道(HOMO)和最低空轨道(LUMO)示意图,见图4。其HOMO和LUMO能量分别为-2.688 9eV和-1.602 4eV,其前线轨道能级差为1.082 5eV。EHOMO和ELUMO均为负值,且能极差较小,在外界刺激下易于发生反应,感度较高;且分子内不含有铅、氯等有毒元素,为潜在的绿色起爆药。
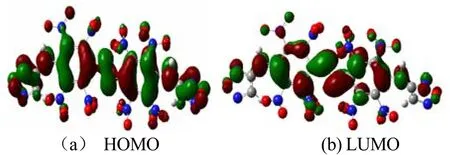
图4 BNFDAONAB分子最高占有轨道和最低空轨道图Fig.4 HOMOandLUMOofBNFDAONAB
2.4静电势
分子静电势(MESP)是分子内静电相互作用力存在的根源之一,其在研究分子间相互作用、反应部位以及分子识别等方面都有着非常独到的作用。有研究证明[14],在含能材料的静电体系中,正的静电势分布的区域联成一片且面积大于负的静电势,本文在6-31+G**基组水平下获得了BNFDAONAB的静电势示意图,如图5所示。图5中蓝色区域为正的静电势,橘红色区域为负的静电势,由图中可见,正的静电势主要分布于偶氮联苯环周围且联成一片,负的静电势主要分布于与环相连的硝基的氧原子周围且面积较小,这与Klaptke[14]等人对含能材料的定性研究相符合,为潜在的含能材料。

图5 BNFDAONAB分子静电势图Fig.5 ElectrostaticpotentialdistributionofBNFDAONAB
2.5密度与生成热
密度和生成热是直接影响含能材料爆轰性能的重要参数,一般认为性能优良的高能量密度化合物(HEDC)其密度应大于1.9g/cm3,爆速应大于9km/s,爆压应大于40GPa[15],6-31+G**基组优化水平下,通过Monte-Carlo方法计算获得BNFDAONAB的体积为384cm3/mol,进而计算获得其理论密度为2.08g/cm3。
等键反应是计算化合物生成热的一种较为精确的方法,因为在等键反应体系中键的类型和数目相同,产物分子和反应物电子环境相似,故电子相关的造成的误差可以相互抵消,使得计算生成热的误差大大降低[10]。本文设计的等键反应如下:

298K条件下,目标化合物的反应热可表示为:

式(1)中:ΔHf,p和ΔHf,r分别代表反应物和生成物在298K时的生成热,NH3、NH2NO2等生成物的生成热均由手册和文献查得,最终确定ΔH298=865.79 kJ/mol。
2.6爆轰性能
爆速(D)、爆压(P)和爆热(Q)是HEDC最重要爆轰特性参数,Kamlet-Jacobs公式是估算分子内只含C,H,O,N元素类含能材料爆轰性能最为常用且较为准确的方法,对于CaHbNcOd类含能化合物,其爆速、爆压和爆热等爆轰参数可由下列公式计算:
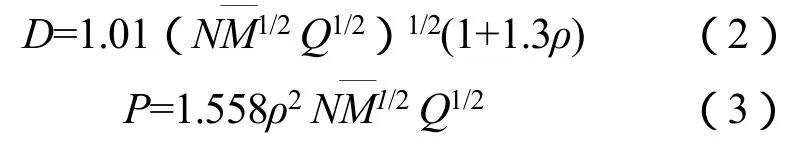
由于该化合物满足b/2c≤c≤2a+b/2,故式(2)~(3)中:

式(2)~(6)中:D为爆速,km/s;P为爆压,GPa;Q为每克炸药爆炸化学能,J/g;——M为气体产物平均摩尔质量;N为每克炸药爆炸产生的气体产物摩尔量,mol/g;M为炸药分子质量;ρ为炸药密度,g/cm3;ΔfHm为炸药的标准摩尔生成焓,kJ/mol。
在一般性能估算中可以使用理论密度代替装药密度,通过计算得到BNFDAONAB的爆速为9.13km /s,爆压为39.03GPa,爆热为4 487.44J/g。由计算结果可知,该化合物的主要爆轰性能的预测值均接近或超过HEDC的要求,综合性能优良,有望获得广泛的应用。
2.7感度性能
撞击感度是表征含能化合物安全性的重要指标之一,早在1979年Kamlet和Adolph就对一些只含C、H、O、N的含能化合物的撞击感度进行了进行了归纳表达,Keshavarz等人[16]在Kamlet和Adolph的研究基础上发现,硝基杂环化合物的撞击感度H50可以通过其元素组成和结构参数进行表达,见式(7)。与试验值比对证明该经验公式的计算值与试验值相符程度较高,通过该公式得到目标化合物的撞击感度H50预测值为1.06cm。从预测的结果来看,BNFDAONAB的撞击感度预测值高于常规制式炸药RDX(42.1cm)和HMX(25.6cm)[8],较为敏感,这也与分子前线轨道预测相一致,BNFDAONAB适合用作高能起爆药。
lgH50=46.29 a'+35.63b'-7.700c'+7.943d'
+44.42n'CNC+102.3n'CNNC(7)
式(7)中:a′、b′、c′和d′分别表示CaHbNcOd炸药分子中C、H、N和O的数目与分子量的比值,n'CNC和n'CNNC是指炸药芳香环中CNC和CNNC部分的数目与分子量的比值。
2.8合成路线设计
采用逆合成分析法,首先考虑官能团的转化,然后完成环与环之间的桥联和偶氮的形成,结合常规的成熟有机反应,目标分子通过图6的合成路线进行。合成所需的主要原料3-氨基-4-硝基呋咱(ANF)早已为多家单位成功合成[17-18],其他原料均为市场销售且价格较为低廉的产品。

图6 BNFDAONAB合成路线Fig.6 Synthetic routesofBNFDAONAB
3 结论
采用密度泛函理论(DFT)的B3LYP方法,在6-31+G**基组水平上对BNFDAONAB进行几何结构全优化并获得其红外光谱,计算过程中所有优化结构无虚频,均为势能面上的真实能量最小点;在优化基础上通过计算获得了其前线轨道能极差、密度和生成热,进而通过Kamlet-Jacobs公式计算获得了其爆速、爆压和爆热;通过其前线轨道能极差和分子静电势分析以及撞击感度预测等方法确定BNFDAONAB是一种感度较高的高能量密度化合物,为一种潜在的绿色起爆药。
[1]Liang Y.H.,Feng J.L.,Xie S.H.,Zhang J.G.,Wang K.,Zhang T.L..Novel high-nitrogen energetic compound based on semicarbazide-substituted tetrazine[J].Chem.Res.Chinese Universities,2012,28(6):931-935.
[2]金兴辉,胡炳成,贾欢庆,吕春绪.3,7-二硝亚胺基-2,4,6,8-四硝基-2,4,6,8-四氮杂双环[3.3.0]辛烷结构与性能的量子化学研究[J].高等学校化学学报,2013,34(7):1685-1690.
[3]Y.Oyumi,T.B.Brill.Thermal decomposition of energetic materials XXVIII.Predictions and results for nitramines of bis-imidazolidinedione:DINGU,TNGUandTDCD[J]. Propellants,Explosives,Pyrotechnics,1988,13(3):69-73.
[4]李战雄.几种呋咱含能衍生物的性能研究[J].含能材料,2005,13(2):90-93.
[5]张淑利,熊贤峰,尉涛,王友兵,王伯周,葛忠学,翟高红,李华.通过红外光谱结合化学计量学方法研究奥克托今的合成机理[J].高等学校化学学报,2012,33(7):1444-1449.
[6]廉鹏,来蔚鹏,王伯周,葛忠学,朱维良,薛永强.新型高能量密度化合物3,6-双(3,5-二硝基-1,2,4-三唑-1)-1,2,4,5-四嗪-1,4-二氧化物的性能预估及合成路线设计[J].化学学报,2009,67(20):2343-2348.
[7]周歌,汪敬,何文娣,田双河,田安民,文忠,赵鹏骥,徐志磊.含能材料六硝基六氮杂异伍兹烷分子的B3LYP研究[J].高等学校化学学报,2002,23(7):1360-1362.
[8]邱玲,肖鹤鸣.由量子化学计算快速预测含能材料晶体密度的简易新方法——HEDM的定量分子设计[J].含能材料,2006,14(2):158.
[9]Fan X.W.,Ju X.H.,Xiao H.M.Density functional theory study of piperidine and diazocine compounds[J].J.Hazard. Mater.,2008,156(1):342-347.
[10]Chi W.J.,Li L.L.,Li B.T.,Wu H.S.Density functional calculations for a high energy density compound of formula C6H6-n(NO2)n[J].J.Mol.Model.,2012,18(8):3695-3704.
[11]Kamlet M.J.,JacobsS.J.Chemistry of detonations.i.A simple method for calculating detonation properties of C-H-N-O explosives[J].J.Chem.Phys.,1968,48(1):23-35.
[12]来蔚鹏,廉鹏,王伯周,罗义芬,二硝基吡唑并吡唑性能的量子化学研究[J].计算机与应用化学,2007,24(8):1025.
[13]肖鹤鸣,陈兆旭,四唑化学的现代理论[M].北京:科学出版社,2000.
[14]HammerlA.,KlaptkeT.M.,NthH.,WarchholdM.,HollG.,Synthesis,structure,molecular orbital and valence bond calculations for tetrazole azide,CHN7[J].Propellants Explos. Pyrotech.,2003,28(4):165-173.
[15]许晓娟,邱玲,肖鹤鸣.高能量密度材料分子设计--基于量子化学计算爆速和爆压[J].含能材料,2004(增刊):505-508.
[16]Keshavarz,M.H.,Pouretedal,H.R.,Semnani,A.Novel correlation for predicting impact sensitivity of nitroheterocyclic energeticmolecules[J].J.Hazard.Mater,2007,141(1):803-807.
[17]李洪珍,李金山,黄明.氨基呋咱氧化为氨基硝基呋咱的合成研究[J].有机化学,2009,29(5):798-801.
[18]张君启,张炜,朱慧,王春华,王有为.一种改进的3-氨基-4-硝基呋咱合成方法[J].含能材料,2007,15(6):577-580.
Studyon QuantumChemistry of Structure andProperties of A Novel High Energetic Density CompoundBNFDAONAB
JING Su-ming,LIU Yu-cun,LIU Deng-cheng,GUO Jia-hu
(SchoolofChemicalEngineering and Environment,NorthUniversityofChina,Taiyuan,030051)
Anovel highenergeticdensitycompound N,N′-Bis(4-nitrofuzan-3-yl-)-4,4′-diamino-2、2′,3、3′,5、5′,6、6′-octanitroazobenzene(BNFDAONAB)was designed.The stable geometry was completely optimized at B3LYP/6-31G+G** theoretical level of density functional theory(DFT),and its IR spectrum was obtained.The heat formation of 865.79kJ/mol and theoretical density of 2.08g/cm3were obtained via isodesmic reaction and Monte-Carlo method,respectively.The detonation velocity,detonationpressure and explosionheatwere predictedbythe formula of Kamlet-Jacobs,based onthe theoretical density. The impact sensitivity was predicted by the formula of Keshavarz.In addition,the reaction route was design by a retro synthesis analysis method.The results showed that BNFDAONAB could meet the requirements of high energetic materials with detonation velocity of 9.13km/s,detonation pressure of 39.03GPa and explosion heat of 4 487.44J/g,predicted value of H50was 1.06cminaddition.Thestudy showed thatthecompound wasapotentialgreenprimary explosive.
Primary explosive;Green;BNFDAONAB;Densityfunctionaltheory;Detonationproperty
TQ560.1
A
1003-1480(2015)02-0039-05
2014-12-15
荆苏明(1986-),男,在读博士研究生,主要从事含能材料合成及性能研究。基金项目:国家自然科学基金委员会和中国工程物理研究院联合基金(No.U1330135)。
猜你喜欢
南昌大学学报(理科版)(2021年4期)2021-11-22
火炸药学报(2020年5期)2020-10-27
食品与健康(2019年7期)2019-07-18
杭州(2019年11期)2019-04-12
火炸药学报(2018年4期)2018-09-01
健康博览(2018年7期)2018-01-03
兵器装备工程学报(2017年10期)2017-11-14
固体火箭技术(2016年2期)2016-11-03
饮食科学(2014年5期)2014-06-18
右江医学(2014年1期)2014-03-22
