
氧化型钙/钙调素依赖性蛋白激酶Ⅱ
2015-10-21 20:00孙胜男吕菁君魏捷
中华急诊医学杂志 2015年3期
孙胜男 吕菁君 魏捷
DOI:10.3760/cma.j.issn.1671-0282.2015.03.026
基金项目: 国家自然科学基金(81372020); 湖北省卫生厅青年科技人才项目(QJX 2012-11);武汉市首批中青年医学骨干人才培养项目
作者单位:430060 武汉,武汉大学人民医院急诊科
通信作者:吕菁君 ,Email: lvjingjun@gmail.com
钙/钙调素依赖性蛋白激酶家族(Ca2+/calmodulin-dependent protein kinase, CaMK)包括CaMKⅠ,CaMKⅡ,CaMKⅢ,CaMKⅣ。其中CaMKⅡ在体内广泛分布,在心肌细胞中高表达。钙/钙调素依赖性蛋白激酶Ⅱ(CaMKⅡ)是多功能的丝氨酸/苏氨酸蛋白激酶,作为Ca2+信号转导的关键因子,调控细胞的多种生物学功能,包括氨基酸和脂质代谢,离子通道/受体及神经递质的合成与释放,维持细胞内钙稳态,调控兴奋收缩耦联作用。
近年来,蛋白质翻译后修饰与心血管疾病的发生发展机制之间的内在联系受到关注。氧化型Ca2+/钙调素依赖性蛋白激酶Ⅱ的发现将Ca2+与氧化应激紧密的联系在一起,NADPH氧化酶/CaMKⅡ 信号通路在心肌梗死后心室重构、病态窦房结综合征的发病机制中起关键性致病作用。氧化型钙/钙调素依赖性蛋白激酶Ⅱ的激活位点、激活通路以及下游效应产物成为新的关注点。
1 CaMKⅡ 蛋白翻译后修饰
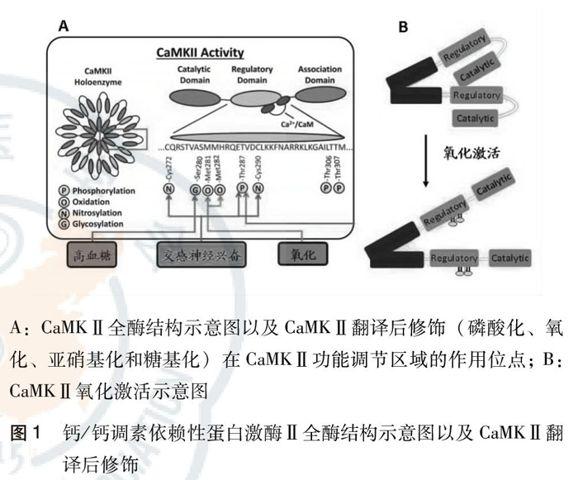
钙/钙调素依赖性蛋白激酶Ⅱ(Ca2+/calmodulin-dependent protein kinase Ⅱ, CaMKⅡ)在20年前被确定是由Ca2+/ CaM 依赖激活的,其磷酸化位点是 CaMKⅡ蛋白活性调节区域第287苏氨酸(Thr287)。研究表明,在没有Ca2+/ CaM 参与下,血管紧张素Ⅱ诱导 CaMKⅡ蛋白活性调节区域第281和第282双蛋氨酸结构氧化,形成氧化型CaMKⅡ(oxidized-CaMKⅡ),导致心肌细胞凋亡[1-2]。进一步的研究表明,CaMKⅡ 第281 / 282 双蛋氨酸结构氧化激活(oxidized-CaMKⅡ)与CaMKⅡ第287苏氨酸位点磷酸化激活(phos-CaMKⅡ)是完全独立的两个过程,并不相互依赖(图 1)。
研究表明,CaMKⅡ的激活存在多种形式,不仅有Thr287磷酸化激活、第281和第282双蛋氨酸氧化激活,糖尿病,长期高血糖导致CaMKⅡ蛋白活性调节区域第280丝氨酸(Ser280)糖基化;交感神经兴奋、神经内分泌过度不仅磷酸化激活第287苏氨酸(Thr287),还导致第272半胱氨酸(Cys272)和第290半胱氨酸(Cys290) 硝基化。
2 CaMK Ⅱ在心脏中的激活通路
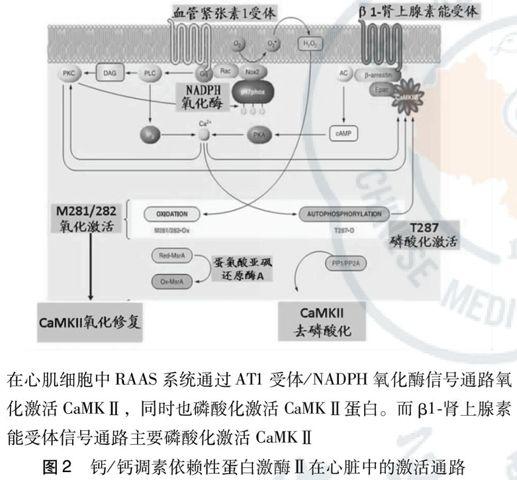
钙/钙调素依赖性蛋白激酶Ⅱ在心脏中的激活通路主要通过肾素-血管紧张素-醛固酮系统(RAAS)和β-肾上腺素能受体信号通路(图2)。在心肌细胞中RAAS系统通过AT1受体/NADPH氧化酶信号通路氧化激活CaMKⅡ,同时也能导致磷酸化激活CaMKⅡ蛋白 [3-5]。而β1-肾上腺素能受体信号通路主要磷酸化激活CaMKⅡ [6-8]。
还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶是体内活性氧(ROS)的最主要的来源之一。NADPH氧化酶的经典结构由6个不同的亚单位组成,它们分别是位于细胞膜的核心亚基gp91 phox和调节亚基p22phox,二者形成的膜复合体称为黄素细胞色素b558,以及位于胞质中的水溶性调节亚基Rac、p67phox、p47phox、p40 phox等。在NADPH氧化酶激活时,胞质中的调节亚基转位与黄素细胞色素b558结合,含黄素的gp91 phox催化亚基作为电子传递系统,以NADPH为递氢体,催化反应:
NADPH+2O2→NADP++H++2O2-。超氧化物阴离子(O2-)在超氧化物歧化酶的作用下转化成H2O2。O2-和 H2O2都是损伤细胞最强的自由基。
血管紧张素激动血管紧张素1受体(AT1受体)诱导NADPH氧化酶活化,细胞内大量的H2O2直接作用于CaMKⅡ蛋白活性调节区域第281/282双蛋氨酸结构,氧化激活CaMKⅡ。激動AT1受体同时也能导致CaMKⅡ蛋白活性调节区域第287苏氨酸磷酸化激活。Mark E Anderson 团队进一步研究证明,异丙肾上腺素通过β1-肾上腺素能受体信号通路,导致CaMKⅡ蛋白活性调节区域第287苏氨酸磷酸化激活,并不作用于第281/282 蛋氨酸[3-5]。
蛋氨酸亚砜还原酶A(methionine sulfoxide reductase A, MsrA)是目前发现的可在体内还原逆转蛋白质蛋氨酸残基氧化结构变化和功能损伤的主要抗氧化酶系统。研究表明,与超氧化物歧化酶(SOD)相比,MsrA不仅可以在细胞内发挥清除氧化因子的作用,还可以对已经发生的蛋白质氧化进行可逆性修复。在心肌细胞内,oxidized-CaMKⅡ第281 / 282 双蛋氨酸结构氧化修复是通过MsrA介导的。在MsrA作用下,心肌细胞内oxidized-CaMKⅡ氧化还原(图2)。过表达MsrA基因可以显著减轻氧化应激引发的心肌细胞损伤和凋亡[9]。
3 phos-CaMKⅡ和oxidized-CaMKⅡ在心血管病理生理中的作用
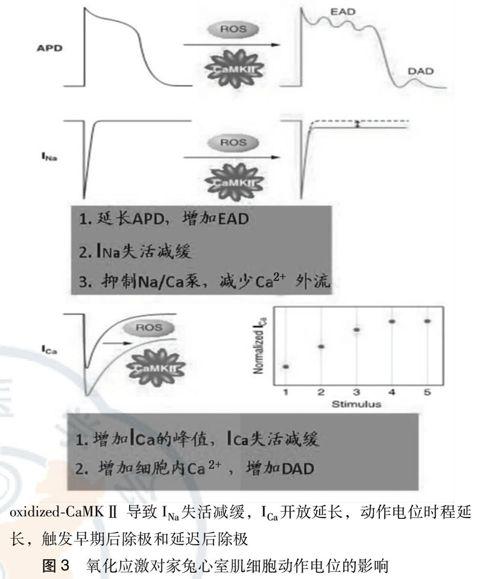
美国加利福尼亞大学洛杉矶分校的Xie等[26]采用膜片钳和钙内流成像负荷技术,研究氧化应激对家兔心室肌细胞动作电位的影响:①由于H2O2氧化激活CaMK Ⅱ,细胞内钙浓度显著增加,触发肌浆网在舒张期不恰当地钙释放,激活瞬时内向电流(Iti)引发延迟后除极。②H2O2(0.2~1.0 mmol/L)作用心室肌细胞5~15 min,影响INa失活,延长动作电位时程。INa通道阻滞剂河豚毒素(10 μmol/ L)可以拮抗这一作用。③ H2O2增强ICa,L的峰值和延长动作电位平台期,增加钙内流,ICa,L阻滞剂硝苯地平可以拮抗这一作用。影响INa失活,扩大ICa,L,延长动作电位时程导致早期后除极现象的发生(图3)。
CaMKⅡ抑制剂KN-93(1 μmol/ L)可以完全拮抗H2O2诱导EADs,无活性的KN-93异构体KN-92无上述作用。选择性CaMKⅡ磷酸化抑制剂AIP (autocamtide-2-related inhibitory peptide)2 μmol/ L也能显著抑制H2O2诱导EADs发作。与CaMKⅡ抑制剂KN-93比较,CaMKⅡ磷酸化抑制剂AIP的抑制作用更多地表现在影响INa失活,CaMKⅡ抑制剂KN-93对INa失活和扩大ICa,L都发挥作用。
Song Y-H 研究证实CaMKⅡ在短暂的氧化压力下诱导大鼠心肌细胞L-型钙电流(ICa,L) 长时程易化(long-term facilitation , LTF) [21]。1 mmol/L H2O2作用大鼠心肌细胞5 min,L-型钙电流长时程易化持续约1 h。CaMKⅡ抑制剂KN- 93能够完全逆转H2O2诱导的L-型钙电流长时程易化。进一步发现,氧化激活CaMKⅡ和磷酸化激活CaMKⅡ都参与维持CaMKⅡ的活化。但是肌浆网Ca2+释放和线粒体
ROS生成在持续CaMKⅡ活化中起关键作用。长时程的心肌动作电位重构是通过氧化CaMKⅡ介导的。线粒体氧化激活CaMKⅡ导致与心律失常相关的病理性心肌细胞记忆。
3.4 oxidized-CaMKⅡ参与心肌缺血-再灌注损伤以及心肌梗死后的炎症反应
CaMKⅡ在心肌中的促炎作用是一个新发现。CaMKⅡ参与心肌缺血-再灌注损伤以及心肌梗死后的炎症反应。Ling等[27]首次研究证实在心肌细胞内CaMKⅡδ直接介导NF-κB激活,CaMKⅡδ触发并维持缺血-再灌注损伤导致的炎症基因表达。CaMKⅡδ基因缺失,减弱缺血-再灌注诱导的炎症反应。基因敲除心脏特异性CaMKⅡδ可以保护心脏,减少缺血-再灌注损伤,减少心肌细胞凋亡,减少梗死面积,改善心脏功能恢复。进一步的研究表明,缺血-再灌注迅速增加CaMKⅡ的活性,在再灌注数分钟内活化NF-κB。活化的CaMKⅡ在心肌细胞中的表达直接导致IκB激酶磷酸化,伴随核P65的增加 。
在心肌梗死中,Toll样受体-4促进核因子κB依赖的炎症反应和氧化损伤。MyD88(myeloid differentiation protein 88)是Toll样受体的重要下游信号和功能蛋白。2012年,Singh等[28] 研究发现脂多糖(LPS)激活Toll样受体-4(TLR-4)诱导心肌细胞CaMKⅡ氧化激活,补体因子B是oxidized-CaMKⅡ激活后的下游效应蛋白。野生型小鼠心肌梗死后心脏中oxidized-CaMKⅡ显著增加,但是MyD88基因敲除(MyD88-/-)小鼠心肌梗死后心脏由于缺乏MyD88依赖的Toll样受体信号激活,不表达oxidized-CaMKⅡ。野生型小鼠心肌梗死后心脏中促炎因子TNF-α、补体因子B、心肌细胞死亡和纤维化均显著增加。与野生型小鼠比较,MyD88-/-小鼠心肌梗死后心肌细胞肥大、凋亡、炎症、oxidized-CaMKⅡ的表达、梗死后病死率均显著减少。原因是MyD88-/- 小鼠核因子κB激活存在缺陷[29] 。这些证据表明,Toll样受体/MyD88这条经典的炎症信号通路氧化激活CaMKⅡ,oxidized-CaMKⅡ活化NF-κB参与了梗死后的炎症反应。
综上所述,作为一个新发现的CaMKⅡ蛋白质翻译后修饰产物,oxidized- CaMKⅡ参与了诸多心血管疾病的发生发展机制。oxidized-CaMKⅡ是肾素-血管紧张素-醛固酮系统和肾上腺素能信号系统的重要效益产物,反映器质性心脏病的氧化应激压力。在心肌细胞中,RAAS系统既氧化激活CaMKⅡ,又磷酸化激活CaMKⅡ。β-肾上腺素能受体信号通路主要磷酸化激活CaMKⅡ。oxidized- CaMKⅡ参与了与心律失常相关的病理性心肌细胞记忆,是病态窦房结综合征患者的生物学标志物。oxidized-CaMKⅡ参与心肌缺血-再灌注损伤,心肌梗死后的炎症反应,以及心肌梗死后醛固酮系统诱导的心室重构。持续的氧化应激压力通过NADPH氧化酶依赖性ROS氧化激活CaMKⅡ,影响INa失活,扩大ICa,L,触发EADs 和DADs,导致器质性心脏病患者发生致命性心律失常。
参考文献
[1]Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKⅡ by methionine oxidation [J]. Cell,2008,133 (3):462-474.
[2]Anderson ME. Pathways for CaMKⅡ activation in disease [J]. Heart Rhythm,2011, 8(9):1501-1503.
[3]Erickson JR, He BJ, Grumbach IM, et al. CaMKⅡ in the cardiovascular system: sensing redox states [J]. Physiol Rev,2011, 91(3):889-915.
[4]Burgoyne JR, Mongue-Din H, Eaton P,et al. Redox signaling in cardiac physiology and pathology [J]. Circ Res,2012,111(8):1091-1106.
[5]Swaminathan PD, Purohit A, Hund TJ, et al. Calmodulin-dependent protein kinase Ⅱ: linking heart failure and arrhythmias [J]. Circ Res,2012, 110(12): 1661-1667.
[6]Lü JJ, Wei J, Lin GS, et al. Effects and mechanism of esmolol given during cardiopulmonary resuscitation in a procine ventricular fibrillation model [J]. Resuscitation, 2009, 80(9): 1052-1059.
[7]吕菁君,魏捷,林国生. 艾司洛尔在心肺复苏中的作用及机制研究[J].中华急诊医学杂志,2008,17(3): 260-266.
[8]吕菁君,林国生,赵冬冬,等. 约克猪钙调控蛋白空间异质性与心室颤动机制[J].中华心血管病学杂志,2008,36(4): 355-359.
[9]Prentice HM, Moench IA, Rickaway ZT, et al. MsrA protects cardiac myocytes against hypoxia/reoxygenation induced cell death [J]. Biochem Biophys Res Commun, 2008, 366(3): 775-778.
[10]陈劲进,肖颖彬,刘健.慢性心房颤动对人心房肌钙/钙调素依赖性蛋白激酶Ⅱ表达的影响[J]. 中国病理生理杂志,2005,21(11):2116-2118.
[11]Swaminathan PD, Purohit A, Soni S, et al. Oxidized CaMKⅡ causes cardiac sinus node dysfunction in mice[J]. J Clin Invest,2011, 121(8): 3277-3288.
[12]Heijman J, Voigt N, Wehrens XH,et al. Calcium dysregulation in atrial fibrillation: the role of CaMKⅡ [J]. Front Pharmacol,2014,5:30. eCollection 2014.
[13]柯俊,陳锋,肖幸,等.钙调蛋白激酶Ⅱ抑制剂对肥厚心肌细胞的影响[J]. 中华急诊医学杂志,2012,21(2):151-155.
[14]廖儒佳,曹雯雯,张伟. 蛋白磷酸酶1与钙/钙调素依赖性蛋白激酶Ⅱ在心肌病中的研究进展[J].中国药理学通报,2012,28(12):1629-1631.
[15]郑铭,韩启德,肖瑞平.心脏中不同β-肾上腺素受体亚型的信号体系及其病理生理意义[J]. 生理学报,2004,56(1):1-15.
[16]Couchonnal LF, Anderson ME. The role of Calmodulin kinase Ⅱ in myocardial physiology and disease[J]. Physiology (Bethesda), 2008,23:151-159.
[17]Kashiwase K, Higuchi Y, Hirotani S, et al. CaMK Ⅱ activates ASK1 and NF-kappaB to induce cardiomyocyte hypertrophy[J]. Biochem Biophys Res Commun, 2005, 327(1): 136-142.
[18]He BJ, Joiner ML, Singh MV, et al. Oxidation of CaMKⅡ determines the cardiotoxic effects of aldosterone [J]. Nat Med,2011, 17(12): 1610-1618.
[19]Joiner ML, Koval OM, Li J, et al. CaMKⅡ determines mitochondrial stress responses in heart [J]. Nature,2012, 491(7423): 269-273.
[20]Luo M, Guan X, Luczak ED, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKⅡ [J]. J Clin Invest,2013,123(3):1262-1274.
[21]Song YH, Choi E, Park SH, et al. Sustained CaMKⅡ activity mediates transient oxidative stress-induced long-term facilitation of L-type Ca2+current in cardiomyocytes [J]. Free Radic Biol Med,2011, 51(9):1708-1716.
[22]Zhao Z, Fefelova N, Shanmugam M, et al. Angiotensin Ⅱ induces afterdepolarizations via reactive oxygen species and calmodulin kinase Ⅱ signaling [J]. J Mol Cell Cardiol,2011, 50(1):128-136.
[23]Figtree GA, Rasmussen HH, Liu CC. Oxidative regulation of the Na+ -K+ pump in cardiac physiology and pathology: clarifying the published evidence [J]. Circ Res, 2013, 112(1):e1.
[24]Wagner S, Ruff HM, Weber SL, et al . ROS-activated Ca/calmodulin kinase Ⅱδ is required for late INa augmentation leading to cellular Na and Ca overload [J]. Circ Res,2011, 108(5): 555-565.
[25]Nishio S, Teshima Y, Takahashi N, et al. Activation of CaMKⅡ as a key regulator of reactive oxygen species production in diabetic rat heart[J]. J Mol Cell Cardiol,2012, 52(5):1103-1111.
[26]Xie LH, Chen F, Karagueuzian HS, et al. Induced afterdepolarizations and calmodulin kinase Ⅱ signaling oxidative stress[J]. Circ Res, 2009; 104(1):79-86.
[27]Ling H, Gray CB, Zambon AC, et al. Ca2+/Calmodulin-dependent protein kinase Ⅱ δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κB[J]. Circ Res,2013, 112(6): 935- 944.
[28]Singh MV, Swaminathan PD, Luczak ED, et al .MyD88 mediated in ammatory signaling leads to CaMKⅡ oxidation, cardiac hypertrophy and death after myocardial infarction [J]. J Mol Cell Cardiol,2012, 52(5): 1135-1144.
[29]Pellicena P, Schulman H. CaMKⅡ inhibitors: from research tools to therapeutic agents [J]. Front Pharmacol,2014,5:21. eCollection 2014.
(收稿日期:2014-10-08)
(本文編辑:郑辛甜)
P335-338
猜你喜欢
中国药房(2021年1期)2021-01-26
国外畜牧学·猪与禽(2020年2期)2020-06-11
国外畜牧学·猪与禽(2019年10期)2019-12-17
读书文摘(下半月)(2019年10期)2019-09-10
中国医药导报(2019年7期)2019-05-13
科教导刊·电子版(2018年9期)2018-06-07
分析化学(2018年2期)2018-03-02
分析化学(2017年12期)2017-12-25
商情(2017年38期)2017-11-28
新教育时代·教师版(2016年47期)2017-04-27
