
胰高血糖素样肽—1结构改造的专利技术综述
2015-10-21 19:13夏文静王岩
安徽农业科学 2015年20期
夏文静 王岩


摘要 胰高血糖素样肽1为一类降血糖多肽药物。国内外学者对其结构也一直进行着不断改进。从胰高血糖素样肽1结构改造的总体概况、专利技术发展态势、重点申请人和重点专利技术的角度,详细阐述胰高血糖素样肽1结构改造领域的技术发展,有利于胰高血糖素样肽1相关领域的药物研发、专利申请等,对今后的相关科研或生产工作有所帮助。
关键词 胰高血糖素样肽1;糖尿病;结构;改造
中图分类号 S-03 文献标识码 A 文章编号 0517-6611(2015)20-012-04
Abstract Glucagon like peptide1(GLP1) is a polypeptide drug which has hypoglycemic effect, and there are many studies on the improvement technology of GLP1. This review introduces the trend of development on glucagon like peptide1 from the perspective about the basic situation of structural modification of glucagon like peptide1, the development of relevant patent technology, the important applicants and the important patent technology, and also elaborates the improvement trend of patent technology on glucagon like peptide1, in order to provide some reference for relevant patent examination and relevant scientific research.
Key words Glucagon like peptide1; Diabetes; Structure; Modification
近年来,胰高血糖素样肽1(Glucagonlike peptide1,GLP1)以其独特的作用机制、良好的治疗效果已被2 型糖尿病治疗领域的科研人员广泛关注。GLP1是一种来自肠道L细胞加工分泌的多肽类激素。1983年Bell等在对胰高血糖素原(Proglucagon,PG)的基因序列进行分析时,发现它可以刺激胰岛素分泌而不出现低血糖,因而能够避免糖尿病治疗中常存在的产生低血糖症的危险。
完整的GLP1由37个氨基酸组成,经过两步酶切,分别去除N端6个氨基酸即形成C 端酰胺化,最终生成具有高度活性的GLP1(736)酰胺(也称GLP1片段)。在人体血循环中,GLP1可被二肽酶Ⅳ(Dipeptidyl pepidiase Ⅳ,DPP Ⅳ;CD26)迅速酶解,即特异性识别GLP1N末端第2位丙氨酸(Ala)残基,从肽链N末端第2位丙氨酸(Ala)处切除二肽,使其由具有生物活性的GLP1(736)酰胺水解成无活性的GLP1(936)酰胺,并且加上较快的肾脏清除速率,天然的GLP1仅有2~6 min短暂的循环半衰期,因此需要通过泵采用持续的皮下灌流以维持体内的GLP1功效。但是,由于GLP1具有其他降糖药物无法比拟的优势,如何延长 GLP 1 的血浆半衰期, 同时保持或增强其生物活性成为各国科研人员以及制药公司的研究目标[1-2]。
以涉及胰高血糖素样肽1及其类似物结构改造的专利数据为分析样本,笔者从专利申请的地区分布、时间分布、申请人状况、专利技术发展脉络等角度详细剖析国内外胰高血糖素样肽1结构改造的专利技术的发展状况,并且在此基础上为国内胰高血糖素样肽1的发展提出建议。
1 中国专利申请整体概况
根据数据库收集的文献量、分布特点,对中文和外文数据库进行选择,其中中文数据库选择CNTXT数据库。CNTXT数据库收录了1985 年至今在中国申请的全部专利文献。通过数据提取,将中国专利代码化数据中的说明书及权利要求这两个部分的信息提取出来,形成中国专利全文文本代码化数据库。考虑到GLP1结构改造后半衰期延长或具备长效性等类似的描述属于技术效果的描述,未必被归为专利申请的发明点而出现在权利要求和说明书摘要中,而是仅作为有益效果出现在说明书中,笔者选择CNTXT作为中文数据库。检索涉及的关键词主要为胰高血糖素样肽1、GLP1、长效、半衰期、延长、变长、更长、增长等。检索涉及的国际专利分类号主要为C07K14/605以及A61K38/26,辅之C07K、C12N、A61K、A61P等。由于检索所使用关键词还涉及胰高血糖素样肽激动剂等技术,因而在检索时还通过排除激动剂等关键词以剔除由所产生的干扰[3]。
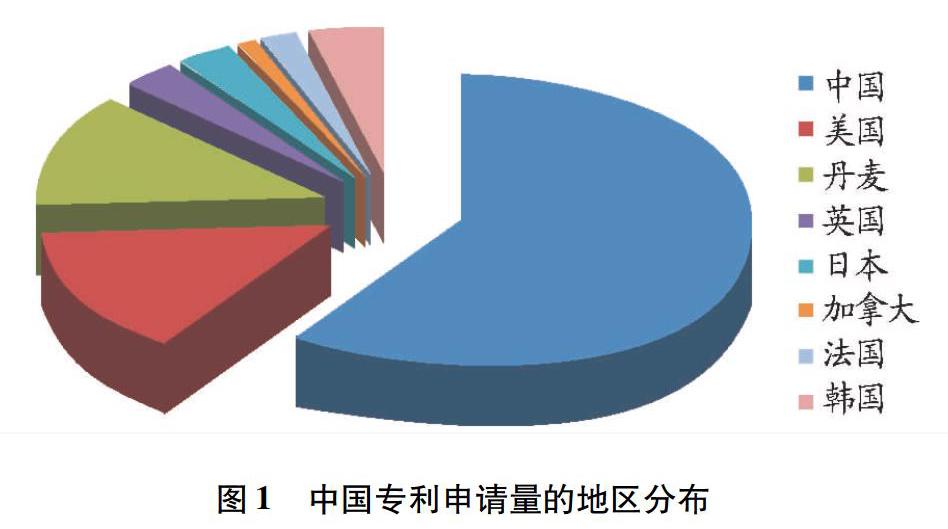
1.1 地区分布
由图1可知,国内申请人的申请量为58件, 占总申请量的58%;国外申请人的申请量为42件,占总申请量的42%,其中美国14件,丹麦12件,这两个国家申请人的申请量合计占国外申请人总申请量的67%,可見美国、丹麦的申请人构成该领域专利申请的主体。
1.2 申请人概况
表1列出了GLP1领域专利申请量排名前四位的国内申请人,即中国药科大学、华东师范大学、暨南大学和天津药物研究院。同时,依托上述科研院所的研究技术,上述高校所在地区的中小型药企的专利申请相对于其他地区较多,如上海、杭州、天津、深圳等地。
表2列出了GLP1领域专利申请量排名前五位的国外申请人。国外申请人的专利申请一般发生于该领域的早中期,对于GLP1的结构改造的研究起不可或缺的作用。以丹麦的诺沃挪第克公司、诺和诺德公司,美国的伊莱利利公司以及后期英国的葛兰素史克等大型制药公司的专利申请为基础,国内高校和公司才逐渐开展该领域的研究改造工作。
2 技术发展脉络
体外化学修饰技术是在多肽水平上,使用合适的修饰方法和修饰剂对其进行化学修饰,使得修饰后的多肽类药物在体内的半衰期延长。目前,国内外研究者主要从两个方面着手进行 GLP1 的结构改造研究(图2)。一是对抗 DPPIV的水解, 主要有GLP1肽链中氨基端的修饰或替换、GLP1肽链 C端的延伸、环化、糖链修饰等;另一方面是延缓 GLP1的肾脏消除速率, 以此为出发点进行的结构改造有聚乙二醇修饰、与大分子蛋白结合、脂肪酸修饰、香豆素修饰以及形成二聚体结构等。
2.1 GLP1肽链中氨基端的修饰、替换或延伸
最早GLP1肽链中氨基端替换的研究始于1990年公开号为JPH05506427 A的文件。在接下来的数十年间,国外的几大公司一直致力于该领域的研究。1995年,美国的伊莱利利公司制备了GLP1类似物包括氨基酸替换、氨基或羧基端的修饰、C6C10的酰化,接着1996~1997年丹麦的诺沃挪第克公开了通过在肽的N末端进行修饰,使GLP1肽类似物能够抵抗DPPIV的降解,延长GLP1肽半衰期的一个备选方法是衍生化作用,其中将阻止DPPIV 接近肽N末端的大的酰基以及亲脂性取代基连接在GLP1的多种氨基酸上。1999年,加拿大的康久化学公司提出一种经修饰的促胰岛素活化衍生物,含有与血液中的氨基、羟基或巯基反应形成稳定共价键的反应性基团。该基团选自琥珀西安亚氨基和马来酰亚胺基,使得该衍生物与血液蛋白上的巯基反应。2001年,美国的伊莱利利公司研发了延伸的C末端偶联的多个位点上具有修饰的GLP1肽,所述修饰提供了增强的稳定性,且其与人白蛋白形成融合蛋白。在接下来一年,该公司又对延伸的C末端偶联的多个位点上具有修饰的GLP1肽进行进一步的改造,以增强其稳定性。同年,日本株式会社三和化学研究所设计了一种GLP1衍生物。它可以通过用丝氨酸取代GLP1氨基酸序列的8位而具有对二肽酶IV抗性,或者分别通过用谷氨酰胺和天冬酰胺取代26位和34位的氨基端而对胰蛋白酶具有抗性。2004年,丹麦的诺沃挪第克公司将GLP1共价连接于杂环羧基生物等以增加分子的血浆半衰期。2005年至今,该领域的专利申请数量开始增加。2005年,该领域的专利申请还以国外公司为主,国内的暨南大学、东北师范大学开始该方面的研究。暨南大学对5、6、8、34、35和5、6、34、35、37位进行修饰,得到半衰期延长的GLP1类似物,而东北师范大学则将GLP1与人胰高血糖素受体或胰高血糖素家族中任何多肽受体及人免疫球蛋白Fc部分、免疫球蛋白Fc部分片段融合。在国外方面,丹麦的诺和诺德在2篇专利中分别提供了一类GLP1类似物,在7、8位上具有至少一种非蛋白质源性氨基酸残基修饰,26或37、38位赖氨酸残基处用部分进行酰化,且其中所述的部分包含至少2个酸性基团,其中一个酸性基团在末端连接。生物相容英国公司和美国的塔夫茨大学信托人也基于GLP1的肽链这氨基端替换或修饰进行深入的研究。在2006~2008年,国内研究还是延续之前构思,如华东师范大学在37位后添加2~3个特定种类的氨基酸,大连帝恩生物工程有限公司将酶解关键性位点突变再与人白蛋白相连,而美国的白时美施贵宝公司、丹麦的诺沃挪第克则对GLP1的N端进行复杂的化学修饰以具备二肽肽酶IV抗性。2008年,中国药科大学加入到GLP1类似物改造的队伍中。他们于8、9、16、22、27或37位点进行改造得到具有更长药理作用时间的GLP1类似物。丹麦的西兰制药公司均对GLP1的N端、C端以及特定位点进行改造,以期具有长效性。2009年,华东师范大学大力进行该领域的研究,提出4篇申请,主要涉及特定位点的改造,而国内中小型药企也开始仿制药的研发,主要涉及相关位点的氨基酸种类替换,在这一年值得一提的是申请人南开大学。它不仅如前将涉及相关位点的氨基酸种类替换,而且将GLP1基因串联表达,达到稳定、长效的生理效果。2010~2013年,该领域的专利申请达20件之多,其中多数是国内高校和中小型药企,研究的方向多是通过氨基端种类的替换以达到增加体内半衰期的目的。
2.2 环化
在多数情况下,机体内的氨肽酶及羧肽酶很容易从常见的直链肽的两端进行逐步地切割分解,使得直链肽被降解。 因此,对肽链进行环化结构改造,可以提高多肽类药物在机体内的生物稳定性,延长半衰期 。按照桥头环化位置,可将环肽分为 C端与N端相连、 侧链间相连、N端与 侧链相连、C端与侧链相连以及主链上的 NN相连5类。2011年,国内的两家制药公司研发了环化的GLP1分子。其中杭州中肽生化有限公司设计的环化GLP1为N端与侧链相连且侧链间相连的环化多肽,而天津拓飞生物科技有限公司开发的为C端与N端相连的环化GLP1。上述化合物有效地延长了药效时间。
2.3 聚乙二醇修饰
聚乙二醇 (Polyethy lene glycol ,PEG )修饰又称分子的 PEG化。 GLP1经 PEG化后相对分子质量增大,肾小球滤过作用减慢,血浆半衰期延长。GLP1分子中有His7、Lys26和 Lys343个位点可进行 PEG修饰。目前应用最普遍的PEG修饰剂是单甲氧基聚醇(mPEG)。当PEG修饰多肽类药物时,为了使PEG能在较温和的反应条件下与多肽偶联,需先将PEG活化, 再将活化后的 PEG与多肽肽链的 C端、N端以及侧链上的氨基、巯基和羧基进行偶联。
该方面申请最早于2003年,专利CN1832959A提供了偶联有至少一个聚乙二醇分子或其衍生物的GLP1化合物,其中每个PEG连在肽的Cys或Lys氨基酸或羧基末端上,导致产生与其非PEG化肽相比半衰期延长和清除率减慢的生物活性肽。这些PEG化GLP1化合物和组合物可用于治疗能够通过降低血液葡萄糖、抑制胃和/或肠蠕动和胃和/或肠排空或抑制食物摄取而受益的糖尿病、肥胖症、过敏性结肠综合征和其他疾病。在随后的2004~2013年间,聚乙二醇修饰的GLP1一直是研究的热点。国内最早研究聚乙二醇化GLP1的专利申请CN1676163始于2004年,申请人为华东师范大学。该专利提出,将GLP1与含经过琥珀酰亚胺活化的活性基团的单甲氧基聚乙二醇组成复合物——单甲氧基聚乙二醇-丙琥珀酰亚胺,即mPEGSPA或分枝状单甲氧基聚乙二醇-琥珀酰亚胺,即mPEGNHS组成,两者通过前者的氨基酸游离氨基和后者的琥珀酰亚胺酯键形成的酰胺键连接在一起。所述的单甲氧基聚乙二醇分子量為5~40 kD。接着,2005年美国的伊莱利利公司也提出GLP1聚乙二醇化的技术方案,它制备了与2个聚乙二醇分子偶联的GLP1化合物或其衍生物,产生与未聚乙二醇化的肽相比具有延长半衰期和降低清除率的生物活性肽。在2008年后,国内制药公司开始加入聚乙二醇化GLP1研究的队伍,如扬子江药业、派格生物医药、江苏豪森药业等,而中国药科大学、华东师范大学等长期研究GLP1结构改造的高校也对聚乙二醇化GLP1做出相关研究。值得一提的是江苏豪森药业研制的分枝型PEG修饰的GLP1类似物,指出目前修饰用的PEG有两分枝型、四分枝型等多种结构。分枝型PEG的接头也有多种,如甘油接头、赖氨酸接头等。对于每种具体结构的生物肽,哪种PEG修饰方式最佳并不固定,而研究发现两分枝的PEG在修饰该发明肽链上具有突出优势。所述分枝型PEG分子量优选40 kD或60 kD,分枝型PEG的接头优选赖氨酸(Lys)。
尽管PEG化聚乙二醇有诸多优势,但PEG修饰剂本身的分散性造成修饰后的药物具有一定的分散性。 这会直接影响产品的生物相容性、 化学性能以及临床功效等。 这也是目前困扰 PEG修饰的一大难点。
2.4 与大分子蛋白结合
在目前的许多方法中,把一个生物活性蛋白(这个蛋白在给药以后易被清除)和一个血浆蛋白(这个蛋白是自然存在的,具有低的清除率)偶联在一起,是一种有前景的提高生物活性肽半衰期的策略(Sheffield WP,Cardiovacs Haematol Disord 1,15,2001)。这种融合蛋白临床应用的优势是能够减少药物注射的频率,提高药物在体内的浓度。一些病原体为免遭排斥而进化获得特异性地结合到免疫球蛋白、白蛋白、纤维结合蛋白或纤维蛋白素原等循环的蛋白质衍生物分子。以上策略与这种病原体的策略相似。
人血清白蛋白(HAS)是血浆中含量最多的蛋白质,浓度约为600 μmol/L,占血浆总蛋白的40%~60%。它也是人体血浆中主要的药物运输蛋白,在人体内的半衰期可达19 d。与人血清白蛋白结合能有效地改善某些肽分子和蛋白药物代谢动力学特性,延长其半衰期。因此,将GLP1与血清白蛋白亲和结合序列融合。该融合蛋白与血清白蛋白亲和结合,可以达到缓释的目的,并且不易被肾脏清除,从而延长GLP1血浆半衰期。
早在2000年,一直致力于GLP1结构改造的美国伊莱利利公司提出将GLP1与人白蛋白融合。接着,在2003年,丹麦的诺沃挪第克公司继其1999年脂肪酸修饰GLP研究后也开始此方面研究,也是将GLP1类似物与白蛋白连接。从2006年开始,该领域成为GLP结构改造的研究热点,涌现出一批专利申请。2006年的GLP与大分子蛋白结合的研究还是集中在GLP1与血清白蛋白结合方面。2007年韩国韩美药品工业株式会社首先提出将GLP1与免疫球蛋白Fc区域相连。在此之前,国际专利公开第WO02/46227号申请,描述了使用基因重组技术通过将GLP1、exendin4或其类似物与人血清白蛋白或免疫球蛋白区(Fc)结合来制备融合蛋白。美国专利第6、756、480号描述了通过将甲状旁腺激素(PTH)和其类似物与Fc区结合来制备Fc融合蛋白。这些方法可以解决如聚乙二醇化产量低和非特异性的问题,但是它们仍具有这样一个问题,即增加血液中半衰期的效果不像期待中的那么显著,并且有时浓度也很低。为了使增加血液中半衰期的效果最大化,使用各种类型的肽连结器,但是可能引起免疫反应。 在上述韩美药品工业株式会社的专利申请CN 101646451中,发明人将免疫球蛋白Fc和非肽聚合物与非N末端的氨基酸残基处通过共价键连接到促胰岛素肽的特异性位点,发现缀合物具有显著地增加体内功效和半衰期的用途。到2008年,国内才出现关于此领域的专利申请。申请号为200810028614.5、名称为“一种缓释生物活性多肽的方法与应用”的发明专利申请提供了一种融合多肽,其中的血清白蛋白结合肽可为LPHSHRAHSLPP,连接体为FNPRGA、FNPRGS、FNPRGP、FNPRPP、FNPRPA,其可以于人血清白蛋白(HAS),从而延长其半衰期。2009年,葛兰素史克的专利WO2011039096以及丹麦的诺沃挪第克公司依然是将GLP1结合于白蛋白上,但是它们均对GLP1的结构进行修饰,使得相应的半衰期延长。2010年,专利CN102533655(申请人青岛黄海制药有限责任公司)虽然研究的仍是GLP/Fc,但是公开了一种高效表达人源GLP/Fc的CHOS细胞株。一直致力于该领域研究的天津药物研究院则提出了一种融合蛋白,即GLP1连接肽GLP1连接肽GLP1连接肽人源蛋白连接肽GLP1连接肽GLP1连接肽GLP1(I),其中连接肽为GGGGS,人源蛋白选自HSA或IgG的Fc片段。2011年,申请人韩美药品工业株式会社在之前研究的基础上,为了克服体重增加、恶心和呕吐的问题,尝试进行长效exendin4缀合物和长效GLP1缀合物的同时施用,同时刺激胰高血糖素样肽1受体和胰岛素受体。结果表明,上述药物同时施用改善了体内功效持续时间和稳定性,并且显著减小了药物的剂量,产生了稳定的血糖水平。另外,他们还发现它改善了由胰高血糖素样肽1激动剂和exendin4或其衍生物诱发的不良事件诸如呕吐和恶心,并且使用长效exendin4缀合物减少了由使用胰岛素所导致的体重增加。 2012年,中国药科大学的黄文龙提出5篇关于GLP1修饰的申请,修饰后的GLP1类似物通过引入小分子基团,增加肽链与血清白蛋白的结合,避免了GLP1的肾脏快速滤过和代谢失活,从而显著延长半衰期及体内降糖作用时间。同年,申请号为200810028614.5的发明人暨南大学的李弘剑提出前次专利所提供的GLP1融合多肽在糖尿病动物模型db/db小鼠中的降糖试验,发现其降糖效果仍有提高的空间,因而对连接体进行改造,从而将其半衰期延长3倍。
2.5 脂肪酸修饰
用脂肪酸修饰 GLP1,可促进其与血浆蛋白 (如清蛋白)结合,延缓肾小球对 GLP1的滤过作用, 从而延长血浆半衰期。脂肪酸修饰GLP1的技术始于GLP1结构改造的早期。1999年丹麦的诺沃挪第克公司的WO9808871A1专利便开始该方面的研究,制备一种GLP1衍生物,其中母体肽的至少一个氨基酸残基连接有一个亲脂性取代基,并且如果只有一個亲脂性取代基,而该取代基又连接到母体肽的N末端或C末端氨基酸残基上,那么该取代基是烷基或具有一个ω羧基的基团。这些衍生物具有比GLP1(737)更长时间的作用曲线和更低的清除率。而丹麦诺和诺德公司开发的Liraglutide(中文名为利拉鲁肽)将 GLP1的 Lys34替换成 Arg34, 并在Lys26残基上连接 16碳酰基链,从而在保留 GLP1活性的同时克服了其易降解的缺点。Liraglutide的半衰期为 11~15 h,仅需每日口服一次就能起到良好的降糖效果。
2.6 香豆素修饰
天然产物中的4羟基香豆素类化合物有较强的血清蛋白结合率。血清白蛋白结合以后的药物与游离药物在体内产生平衡,缓慢释放,实现长效化。同时,血清白蛋白结合药物不被肾小球滤过,可以避免肾脏的滤过代谢。依据上述构思,2011年中国药科大学黄文龙等将4羟基香豆素为母核的香豆素类化合物,通过半胱氨酸的巯基与GLP肽链相连接,并对肽链中相关氨基端修饰,从而制备了一种胰高血糖素样肽1(GLP1)类似物。它在保留降糖活性的基础上,具有抗肾脏滤过消除和抗DPPIV酶解作用,生物半率期较GLP1原型长,提高GLP1的稳定性,延长作用时间。
2.7 糖链修饰
与未聚乙二醇化(unPEGylated)肽相比,经过聚乙二醇修饰可得到半衰期延长、清除延迟化的的GLP1。但是,PEG为在生物体内不代谢的化合物,因此在持续给药聚乙二醇化GLP1化合物时,存在PEG蓄积在生物体内、对生物体产生药害的危险性。糖链在生物体内担负着多种重要生理功能。虽然研究者已意识到其重要性,但因其结构的复杂性、多样性而导致研究比较落后。多肽药物的糖基化修飾可以增加侧链的空间位阻, 提高多肽对酶的稳定性,如LY2189265是一种新型的长效胰高血糖素(GLP1) 受体拮抗剂, 用于治疗 Ⅱ型糖尿病。通过糖基化修饰, 该药物只需每周注射 1次, 大大提高了病人的依从性。
而在专利研究方面,日本株式会社糖锁工学研究所的梶原康宏等于2007~2009年的3篇专利均涉及GLP1的糖链修饰。与GLP1相比,附加糖链的GLP1肽血中半衰期长,且优选显示高的血糖值抑制活性。上述附加糖链的GLP1肽的特征在于在(a)GLP1、(b)GLP1中缺失、取代或附加1或数个氨基酸的肽或在(c)GLP1的类似物中至少1个氨基酸被附加糖链的氨基酸取代,一般为附加糖链的Asn或附加糖链的Cys,其中糖链和氨基酸可以通过接头结合,也可以不通过接头结合。糖链一般优选为由4个以上糖构成的糖链。种类选自二唾液酸糖链、单唾液酸糖链、去唾液酸糖链、二N乙酰葡萄糖胺糖链及二甘露糖糖链的糖链等。
2.8 形成二聚体结构
为抵抗二肽酶IV对GLP1的降解,延长在体内的药效时间,已有多种方法,如上述氨基酸的定点突变、分子修饰等,但是改造后的分子与天然GLP1的同源率低,差异较大,导致不同程度的副作用如过敏、呕吐、胃肠不适等。 2011年,华东师范大学劳勋等提出如下对GLP1的改进:在GLP1衍生物DLG3312的结构XYX中,X与Y的连接方式为2个X的碳端羧基分别与Y的2个氨基缩合连接,其中,X表示的序列为HX8EGTFTSDVSSYLEGQAAKEFIAWLVKGRG,其中X8是A、G、dA或V中的任意一个,dA表示DAla;Y表示二氨基羧酸,包括Lys、Orn。所述GLP1衍生物DLG3312通过所述X的碳端羧基与所述Y的氨基缩合连接成同源二聚体,由此形成由2个X与1个Y构成的特定结构的U型同源二聚体。它具有如下优点:与受体的亲和力相对于单体有较大提高,从而能够更有效地激活受体;与单体相比,二聚体具有相对较大的分子量,由此相对延长因肾小球滤过作用对其清除的时间,使之在体内停留时间增加,延长药效时间等。 另外,专利CN103059127、CN103059127也涉及形成二聚体结构GLP1。二聚体的形成在某些程度上极大地延长了其在体内的半衰期。在某些程度上极大地延长了其在体内的半衰期。
3 展望
虽然目前对GLP 1 及其类似物的结构改造研究已取得良好的效果,但也要看到不足之处,如多数药物仍存在不良反应,最常见的是胃肠道反应即恶心、呕吐,艾赛那肽(Exenatide)还可能导致部分患者出现急性胰腺炎。将化学与生物方法相结合应用于多肽药物长效化技术的探索,集两者的优势,共同开发新型的复合型长效化GLP1是将来的趋势。
参考文献
[1]孙灵芝,阮学平,阿丽塔.胰高血糖素样肽- 1及其类似物的结构改造研究进展[J].医学研究杂志,2010,39(7):17-19.
[2] 姜和,龚梦嘉.长效多肽药物化学修饰的研究进展[J].重庆理工大学学报:自然科学版,2012,26(4):33-39.
[3] 冯刚.聚苯醚产业中国专利分析[J].塑料工业,2010,38(9):1-5.
猜你喜欢
中老年保健(2022年5期)2022-08-24
中老年保健(2022年1期)2022-08-17
中老年保健(2021年5期)2021-08-24
中老年保健(2021年11期)2021-08-22
哲学评论(2021年2期)2021-08-22
影视与戏剧评论(2016年0期)2016-11-23
科学与财富(2016年28期)2016-10-14
科学与财富(2016年28期)2016-10-14
