
没食子酸分子印迹聚合物电化学传感器的制备及研究
2015-10-18 03:03李利军白大伟李彦青崔福海
分析科学学报 2015年4期
张 雷, 李利军, 白大伟, 程 昊, 李彦青,崔福海, 赵 斌, 胡 延
(广西科技大学生物与化学工程系,广西柳州 545006)
没食子酸化学名称为3,4,5-三羟基苯甲酸(Gallic Acid,GA),是可水解单宁的组成部分,又称五倍子酸,它不仅有较强的抗氧化作用,还有较强的抗菌、抗病毒和抗肿瘤作用[1]。目前,有关没食子酸的含量测定方法有薄层扫描法[2]、高效液相色谱法[3]、毛细管电泳法[4]、流动注射法[5,6]和电化学方法[7]等。但这些方法存在灵敏度不高,或选择较差,或操作繁琐耗时等缺点。
分子印迹技术是通过模板分子与功能单体之间的共价键或非共价键作用,在聚合过程中将模板分子固定在交联的聚合物网络上,除去模板分子后留下与模板分子形状相匹配的孔穴,在合成材料中形成具有高亲和力和高选择性的分子识别位点[8 - 10]。分子印迹聚合物(MIP)膜具有选择性强、化学性质稳定、对环境的耐受性好和制备简单、造价低廉等优点[11],与电化学技术相结合,研制的分子印迹电化学传感器不仅选择性较好,而且灵敏度较高。当前有关没食子酸的分子印迹电化学传感器的研究尚少见报道。本文以没食子酸为模板分子,采用自由基热聚合的方式合成MIPs,构建了测定没食子酸的新型分子印迹电化学传感器,并对其性能进行了研究。
1 实验部分
1.1 仪器与试剂
CHI660b电化学分析仪(上海辰华仪器有限公司),三电极系统:玻碳电极或修饰电极为工作电极,213型铂片电极为对电极,232型饱和甘汞电极为参比电极。ZFD-A5040A型恒温干燥箱(北京宏昌信科技有限公司);PE-521)型旋转蒸发仪(上海青浦沪西仪器厂);DF-101S集热式恒温加热磁力搅拌器(上海越众仪器设备有限公司)。
没食子酸(GA,天津市光复精细化工研究所);α-甲基丙烯酸(α-MAA)、偶氮二异丁腈(AIBN)(化学纯,国药集团化学试剂有限公司);乙二醇二甲基丙烯酸酯(EGDMA,阿拉丁试剂);焦性没食子酸、水杨酸、对羟基苯甲酸、水杨酸、KCl、K3[Fe(CN)6]、乙腈、甲醇、HAc等均为分析纯。实验用水为二次蒸馏水。
六味地黄丸(马鞍山天福康药业有限公司)。
1.2 玻碳电极的预处理
将玻碳电极(GCE,Φ=3 mm)依次用0.3、0.05 μm Al2O3悬浊液在鹿皮上打磨抛光,用水清洗干净后,将电极依次置于HNO3(1+1)、无水乙醇和二次蒸馏水中各超声清洗5 min,最后用二次蒸馏水冲洗干净待用。将预处理好的GCE置于0.5 mol/L H2SO4中,在-0.6~+0.8 V电位区间,以50 mV/s的扫描速度扫描20圈,直至循环伏安曲线稳定。
1.3 分子印迹和非印迹修饰电极的制备
取模板分子没食子酸(0.1 mmol),功能单体MAA(0.6 mmol),溶于20 mL乙腈,置于单口烧瓶中,超声振荡5 min,静置2 h,使没食子酸和MAA充分作用,而后加入3 mmol EGDMA和0.01 g AIBN,超声混合均匀。将烧瓶连接旋转蒸发装置,于温度55 ℃下减压蒸馏-沉淀聚合3 h,然后取3 μL上述溶液均匀滴涂在处理好的GCE表面,置于55 ℃恒温干燥箱内再聚合2 h,电极表面形成了一层均匀透明的聚合物膜。将聚合好的分子印迹电极(MIP/GCE)置于含有甲醇-HAc的溶液中浸泡,以除去印迹分子,再用二次蒸馏水洗去印迹电极表面的甲醇和HAc,将其保存于二次蒸馏水中待用。非印迹电极(NIP/GCE)的制备除不加入模板分子外,其余步骤同上。
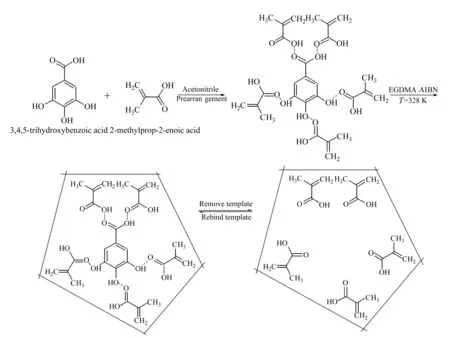
图1 以没食子酸为模板的分子印迹聚合物膜的制备过程示意图
2 结果与讨论
2.1 分子印迹聚合物的制备
分子印迹聚合物的制备过程如图1所示,MAA与没食子酸在乙腈中以氢键(没食子酸上的羟基与MAA上的羰基间,以及没食子酸上的羰基与MAA上的羟基均有可能形成氢键)等非共价键的形式预先自组排列,然后加入交联剂EGDMA交联聚合,使其形成空间网状聚合物,最后在适当溶剂的作用下除去没食子酸,从而在聚合物膜内留下与没食子酸空间大小相匹配并能可逆结合没食子酸的“孔穴”。
2.2 分子印迹膜的性质
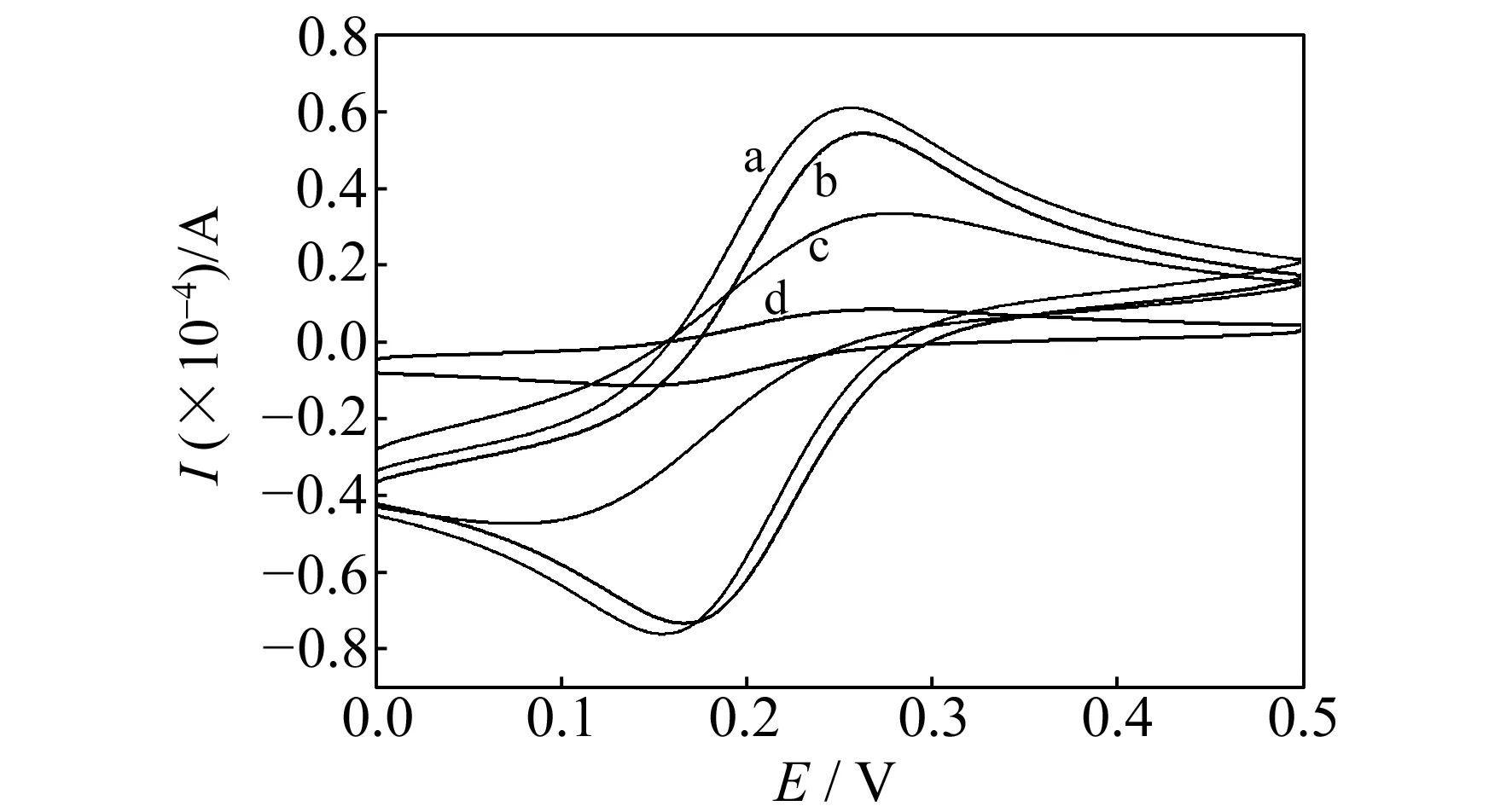
图2 不同电极在K3[Fe(CN)6]溶液中的循环伏安图
图2为不同电极在K3[Fe(CN)6]溶液中的循环伏安曲线。将非印迹电极经甲醇-HAc洗脱后,于K3[Fe(CN)6] 溶液中进行循环伏安扫描,该电极响应很小(图2曲线d),结果表明电极的表面形成了一层较致密的聚合物膜,导致K3[Fe(CN)6]向电极表面的扩散比较困难。将印迹电极于甲醇-HAc中浸泡30 min后,K3[Fe(CN)6]在该电极上的循环伏安曲线的峰电流比在相同条件下处理过的非印迹电极显著增大(图2曲线b),但比裸电极有所下降(图2曲线a),说明印迹分子没食子酸经甲醇-乙酸洗脱后,在印迹膜上形成了匹配的“孔穴”,允许K3[Fe(CN)6] 扩散到电极表面,因此电化学信号增强;但电极表面的大部分还是被印迹聚合膜所覆盖,所以其峰电流比裸电极小。加入一定浓度的没食子酸与印迹电极作用后,峰电流有所下降(图2曲线c),这是由于印迹电极吸附了没食子酸,占据了部分“孔穴”,导致扩散到电极表面的K3[Fe(CN)6] 分子减少。
2.3 模板分子洗脱溶剂的选择
研究了20 mL体积比分别为9∶1、4∶1、3∶2、1∶1的洗脱溶液对模板分子的洗脱效果。实验结果表明,当甲醇和HAc体积比为9∶1时,模板分子去除困难,这是因为洗脱溶剂的极性较弱,难以破坏氢键,所以模板分子难以去除;当甲醇和HAc体积比为3∶2和1∶1时,虽然能迅速的洗脱掉模板分子,但不能维持聚合物膜骨架的稳定,这是因为洗脱溶剂的极性较强,使功能单体与交联剂的聚合能力和交联程度均有所下降,导致聚合物膜稳定性变差;而用体积比为4∶1的甲醇-HAc溶液作为模板洗脱溶剂时,既可迅速去除模板分子,又可保持聚合物膜骨架的稳定性。因此选择20 mL体积比为4∶1的甲醇-HAc溶液作为去除模板的最优溶剂。
2.4 模板分子洗脱时间的选择
用20 mL体积比为4∶1的甲醇-HAc溶液作为洗脱溶剂进行洗脱,洗脱时间与方波伏安氧化峰电流关系的研究结果表明(图3),随着洗脱时间增加,印迹膜电极在K3[Fe(CN)6]溶液中的峰电流值逐渐增大,当洗脱时间达到30 min时,电极在K3[Fe(CN)6]溶液中的峰电流值变化趋于稳定,所以本实验选择30 min作为最佳洗脱时间。
2.5 吸附时间的影响
在含有一定浓度的模板分子溶液中,通过对吸附时间与方波伏安氧化峰电流关系的研究(图4),发现随着吸附时间的增加,印迹电极在K3[Fe(CN)6]溶液中的峰电流逐渐减小,但是当吸附时间大于12 min后峰电流基本不再减小,说明吸附趋于饱和。本实验选择吸附时间为12 min。
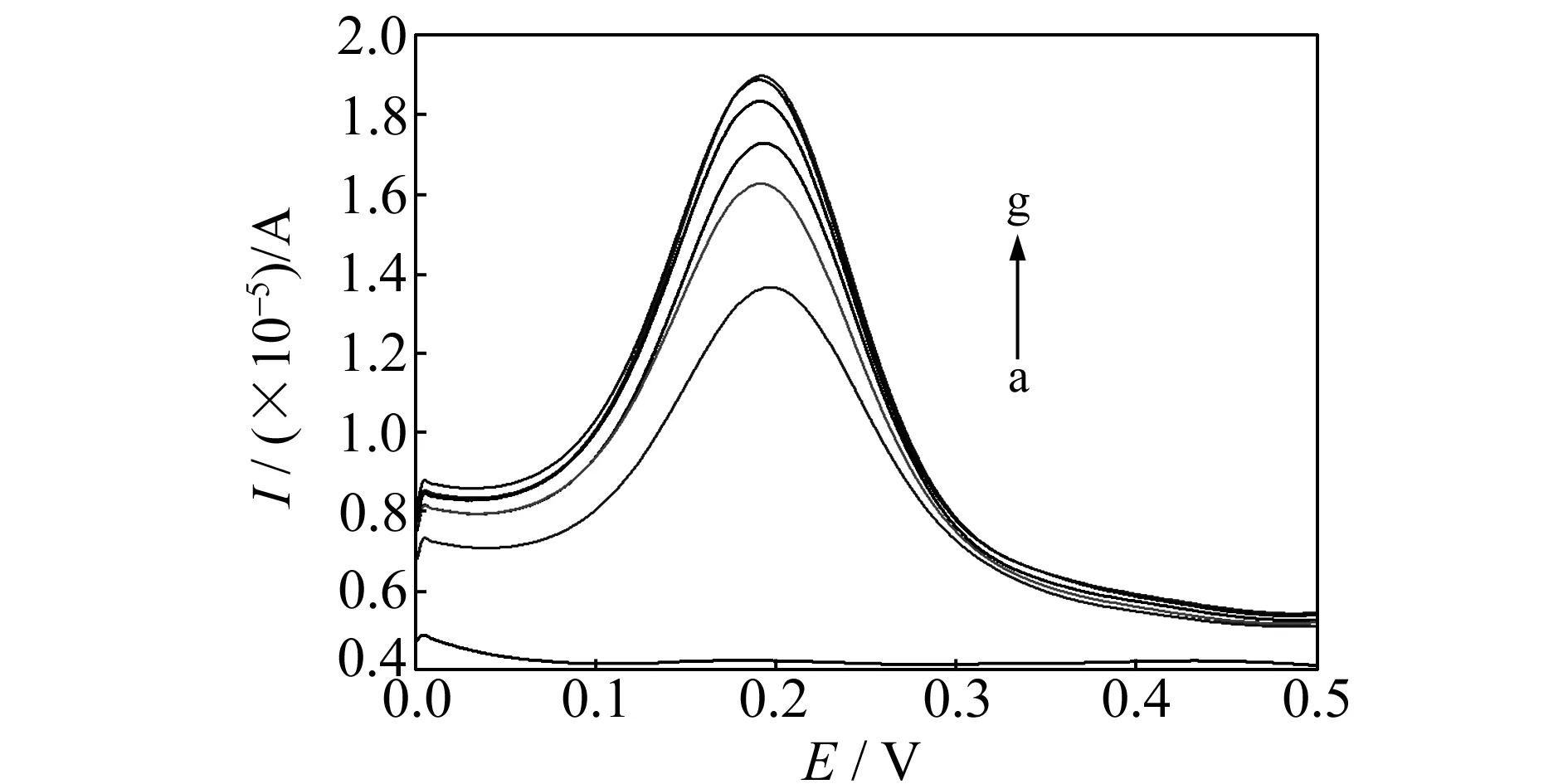
图3 不同洗脱时间的印迹电极在K3[Fe(CN)6]溶液中的方波伏安图
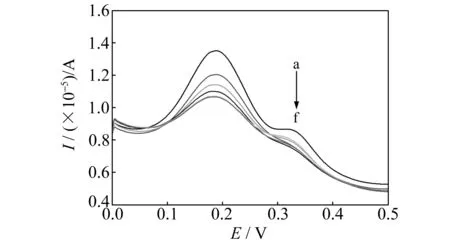
图4 不同吸附时间的印迹电极在K3[Fe(CN)6]溶液中的方波伏安图
2.6 不同浓度的没食子酸与K3[Fe(CN)6] 峰电流的关系
在最优实验条件下,将洗脱完全后的印迹电极在不同浓度的没食子酸溶液中吸附相同的时间,记录K3[Fe(CN)6] 氧化峰电流的变化,结果表明随着没食子酸浓度的增加,K3[Fe(CN)6]氧化峰电流逐渐降低。考虑到电极经过多次处理后电极表面电化学活性存在一定的差异,所以实验用氧化峰电流差△I来描述峰电流的变化,△I为未加入没食子酸时与加入某一浓度的没食子酸时K3[Fe(CN)6]溶液的氧化峰电流之差[12]。结果表明,△I与没食子酸的浓度在4.69×10-6~2.14×10-5mol/L的范围内呈现出良好的线性关系,其线性回归方程为:△I(μA)=0.5047c(μmol/L)-0.8625(R2=0.9962),检出限(S/N=3)为6.41×10-7mol/L。
2.7 传感器的选择性、重现性及稳定性
在选定的最佳实验条件下,固定没食子酸的浓度为1.0×10-5mol/L,考察了其它类似物对没食子酸印迹膜的影响。结果表明,允许误差范围≤±5%,3倍的焦性没食子酸,10倍的水杨酸、对羟基苯甲酸、苯甲酸对没食子酸的测定均不产生干扰,说明该传感器对没食子酸具有较好的选择识别性。
同一支印迹电极对1.0×10-5mol/L的没食子酸溶液平行测定5次,其相对标准偏差(RSD)为3.96%,表明该印迹电极有较好的重现性。同一支印迹电极于4 ℃放置冰箱一周后,对没食子酸的响应值能达到初始值的90%以上,表明该印迹电极有较好的稳定性。
2.8 样品分析及回收率
取六味地黄丸颗粒,研磨粉碎后,加入一定量的乙醇超声溶解,离心后取上清液转至100 mL容量瓶中定容,加标回收平行测定上述溶液三次,结果见表1。
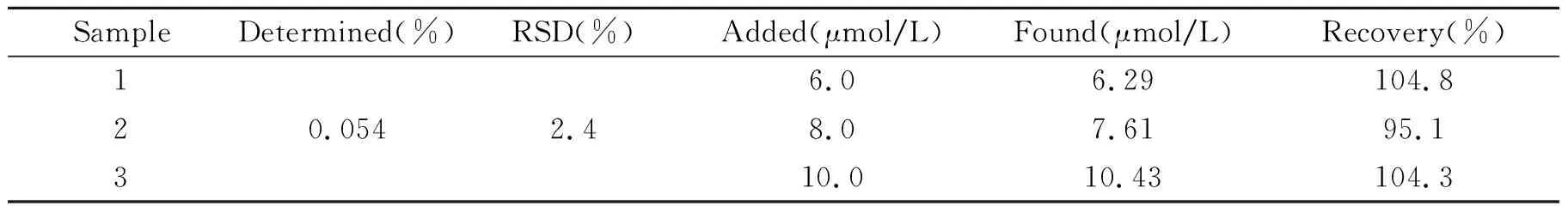
表1 样品分析及回收率试验结果(n=3)
结果表明,该方法所制备的分子印迹电化学传感器对没食子酸的检测具有较好的重现性和较高的准确度,用于六味地黄丸样品的检测,回收率在95.1%~104.8%之间,平均相对标准偏差为2.4%。
3 结论
以没食子酸为模板分子,采用蒸馏-沉淀自由基热聚合法,缩短了聚合所需的时间,成功研制了没食子酸分子印迹电化学传感器。利用方波伏安法,以铁氰化钾为电活性探针,建立了间接测定没食子酸的电化学分析新方法,该方法具有选择性高、检出限低、重现性和稳定性好等特点,用于六味地黄丸中没食子酸含量的检测,结果满意。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
中学生数理化·中考版(2022年12期)2022-02-16
云南画报(2021年10期)2021-11-24
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
表面工程与再制造(2019年6期)2019-08-24
小学生优秀作文(高年级)(2018年4期)2018-09-11
资源节约与环保(2018年1期)2018-02-08
池州学院学报(2017年3期)2017-10-16
电子制作(2016年23期)2016-05-17
肇庆学院学报(2016年5期)2016-03-11
