
铬基催化剂在氟氯交换反应中的研究进展
2015-10-11 05:56张文霞罗建伟王月娟罗孟飞
化工生产与技术 2015年5期
张文霞 梁 艳 罗建伟 王月娟 罗孟飞
(浙江师范大学物理化学研究所,浙江 金华 321004)
氟氯烃(CFCs)曾被广泛应用于制冷剂、发泡剂、净化剂、溶剂、灭火剂、气溶胶推进剂和抛光剂等诸多领域。但由于CFCs分子中的氯原子在强紫外线作用下易裂解形成Cl自由基,而Cl自由基会消耗臭氧,因此CFCs被公认为臭氧层损耗物质。随着《关于消耗臭氧层物质的蒙特利尔议定书》和《中国逐步淘汰消耗臭氧层物质的国家方案》的进一步实施,CFCs替代品的开发变得尤为重要。
在氟化工领域,由于氟代烷烃(HFCs)的臭氧消耗潜能值(ODP)为 0,全球变暖潜能值(GWP)较小,制冷性能与CFCs相似,是CFCs的理想替代品[1-4]。HFCs的合成途径主要有加氢脱氯法和氟氯交换法[5]。由于加氢脱氯法存在催化剂为贵金属催化剂,不仅生产成本高,且活性组分容易流失和烧结、寿命较短、重复使用后再生及回收困难等缺陷,而氟氯交换法具有易于控制、三废污染少、催化剂寿命长、且便于大规模连续生产等优点,因此目前工业上一般采用气相氟氯交换反应路线合成一系列HFCs,如[6-9]:

近年来,氢氟烯烃(HFOs)因零 ODP,更低的GWP也被公认为目前较为理想的CFCs替代品。比如 1,3,3,3-四氟丙烯(HFO-1234ze)和 2,3,3,3-四氟丙烯(HFO-1234yf)不仅环境参数优越(ODP=0,GWP=6),且无毒无味,化学性能稳定,制冷效率高,因此在冰箱、空调制冷剂、气溶胶推进剂和泡沫塑料发泡剂中被广泛应用;而顺-1,1,1,4,4,4-六氟-2-丁烯(Z-HFO-1336)由于含有双键,进入大气后易和OH自由基发生加成,经氧化后降解,所以寿命期气候性能值小,GWP低,又因沸点和室温相近,故其具有良好的发泡和隔热性能。
气相合成氟代烃(HFCs和HFOs)的主体催化剂是铬基催化剂[10-12]。虽然氧化铬具有毒性,但目前尚未找到有效的替代氧化铬的催化剂。因此,目前用于气相氟化合成氟代烃的铬基催化剂主要有铬基本体催化剂和负载型铬基催化剂,催化剂的性能和制备工艺存在密切的关系[13-17]。本文对气相氟化合成氟代烃的铬基催化剂的制备方法,影响铬基催化剂性能的因素以及铬基催化剂失活的原因进行了系统的归纳和分析,并指出了今后铬基催化剂在氟氯交换反应中的发展方向。
1 氟氯交换反应铬基催化剂的制备
1.1 沉淀法
沉淀法尤其是共沉淀法在催化剂的制备中应用比较广泛。将铬的可溶性金属盐或铬与其他元素的可溶性金属盐溶于水中,加入沉淀剂生成沉淀物,经过滤、干燥、粉碎、成型和焙烧等步骤制得催化剂。Wang等报道了Y2O3-Cr2O3催化剂的制备方法及其催化性能。将一定量的Cr(NO3)3·9H2O和Y(NO3)3·6H2O的混合溶液用沉淀剂NH3·H2O沉淀,分离得到的沉淀物经干燥后于600℃氮气气氛下焙烧4 h,制得不同Y含量的xY2O3-Cr2O3催化剂(x为催化剂中Y的摩尔分数)[18]。
在制备过程中,沉淀剂、沉淀温度、溶液浓度、加料顺序、搅拌速度以及pH值等均会影响催化剂的孔容、比表面积、晶形等物理性质。沉淀法的优点是活性组分和载体的结合力强,反应中活性组分流失较少,但由于催化剂的重复再现性较差,因此对实验操作要求较高。
1.2 浸渍法
浸渍法是用铬或其他组分的可溶性盐溶液浸渍载体,采用过滤、蒸发等方式除去过剩的溶液,再经干燥、焙烧和活化制得催化剂。一般可以通过控制浸渍的时间和温度,以及溶液的浓度来调节活性组分在载体表面上的分布。吕剑等用不同含量的CrCl3溶液或 CrCl3和CoCl2的混合溶液浸渍 γ-AlF3载体,制得一系列不同Cr3+含量的催化剂前驱体,再于350℃氮气气氛下进行焙烧,最后经过HF活化后得到 CrF3/AlF3或 CrF3·CoF2/AlF3催化剂[19]。
浸渍法的优点是能将一种或几种活性组分负载于载体上,且催化剂的形状、表面积、孔隙率等主要取决于载体,因此一般选择比表面积较大的载体,如活性炭和改性AlF3[20]。但由于活性组分之间的竞争吸附以及浸渍液溶质的迁移的存在,因此会导致催化剂的活性组分分布不均匀,稳定性下降[21]。
1.3 共混法
共混法可根据混合时原料的状态分为干混和湿混。干混法是将Cr2O3和载体按一定比例混合、研磨和捏合成型,经过焙烧和氟化得到氟化催化剂;湿混法是将载体加入到活性组分的水溶液中,在连续搅拌下滴加NH3·H2O,使活性组分沉积在载体表面,制得相应的催化剂前驱体。罗孟飞等将Cr2O3、Ni(OH)2、AlF3和Ag2O机械混合,压制成型,再经氮气气氛焙烧,氢气气氛还原后制得Ni-Ag-Cr2O3-AlF3催化剂[22]。胥会祥等以碱式碳酸镁和氢氟酸制备MgF2,以CrCl3·6H2O和氨水制备Cr(OH)3·H2O,将2者混合均匀后得到催化剂前驱体,用HF活化后制得Cr/MgF2催化剂。共混法的优点是制备工艺简单,是工业制备氟氯交换催化剂的首选方法[23]。
2 催化剂性能的影响因素
2.1 制备方法的影响
Cho等考察了催化剂制备方法对催化性能的影响,分别采用共沉淀法和浸渍法制备了CrOx/MgO催化剂,并用于气相氟化1,1,1-三氟-2-氯乙烷(HCFC-133a)合成 1,1,1,2-四氟乙烷(HFC-134a)反应中进行研究。结果表明,当反应温度为320℃时,采用共沉淀法制备的CrOx/MgO(共沉淀法)催化剂的转化率为9.8%,而用浸渍法制备的CrOx/MgO催化剂的转化率仅为1.0%,这可能是由于CrOx/MgO(共沉淀法)的比表面积(94 m2/g)远大于CrOx/MgO(浸渍法)的比表面积(10 m2/g)所造成的[24]。
Rao 等比较了 Cr2O3、Cr2O3/Al2O3(浸渍法)和Cr2O3/Al2O3(共沉淀法)催化剂对气相氟化HCFC-133a活性与寿命的影响。结果发现,3种催化剂的活性差别不大,但催化剂的寿命以Cr2O3>Cr2O3/Al2O3(共沉淀法)>Cr2O3/Al2O3(浸渍法)顺序依次降低[25]。
2.2 载体的影响
Cho等在气相氟化HCFC-133a合成HFC-134a研究中发现,催化剂的活性与载体有关,不同的载体会影响催化剂表面Cr物种的价态和分散性,进而影响其催化活性。实验结果表明,在350℃反应温度下,CrOx/MgO、CrOx/Al2O3催化剂的活性明显高于CrOx/MgF2、CrOx/ZrO2催化剂 (实验结果如表 1所示)。这主要是因为只有CrOx/MgF2、CrOx/ZrO2催化剂中存在晶相Cr2O3,而CrOx/MgO、CrOx/Al2O3催化剂中没有观察到晶相Cr2O3,这表明CrOx/MgO、CrOx/Al2O3催化剂中存在高价态的CrOx物种,而高价态的CrOx物种在活化过程中转变成的高分散CrxFy和CrxFyFz是反应的活性物种,因此CrOx/MgO、CrOx/Al2O3催化剂的活性高[24]。
2.3 前驱体的影响
钱林等将一定量的Cr(NO3)3·9H2O分别和Y(OH)3、YCl3·6H2O、Y(NO3)3·6H2O 的混合溶液用沉淀剂NH3·H2O沉淀,制得CrOx-Y2O3催化剂。结果发现在气相氟化合成HFC-134a的反应中,Y前驱体会极大地影响催化剂表面Cr物种价态的分布,其中以Y(OH)3为前驱体制备的CrOx-Y2O3催化剂能促进Cr物种以高分散的CrO3形式存在,而CrO3在预氟化阶段及反应过程中可转化为活性物种CrOxFy和Cr(OH)xFy,因此相比于其他前驱体制备的催化剂,该催化剂的活性更高[26]。
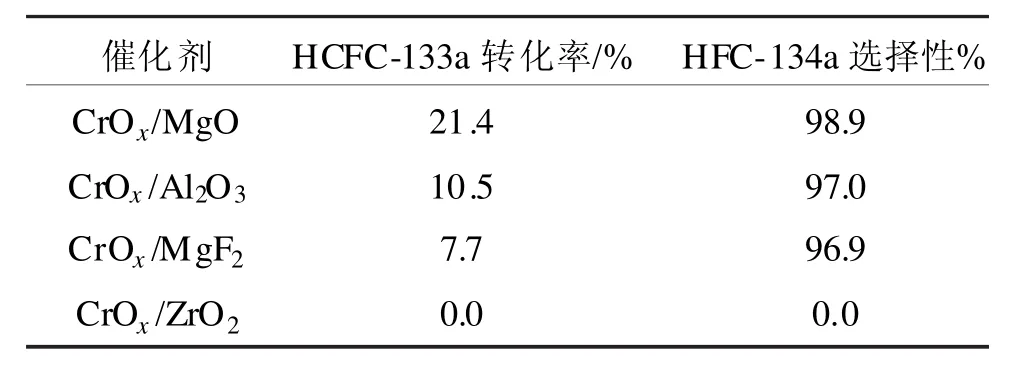
表1 350℃下不同载体负载的催化剂催化氟化HCFC-133a合成HFC-134a的反应结果Tab 1 Reaction results of HFC-134a catalytic fluorination by HCFC-133a use catalyst with different load carrier at 350℃
2.4 铬含量的影响
Jia等制备了不同Cr2O3含量的xCr2O3-AlF3催化剂 (x为Cr2O3的质量分数),并用于气相氟化CCl2F2合成CF4的研究。结果表明,催化剂的活性同Cr氧化物与AlF3间的相互作用有关。和单一的Cr2O3和AlF3催化剂相比,61.2Cr2O3-AlF3催化剂的活性更高[27]。
吕剑等在γ-AlF3上负载了不同含量的Cr3+,以研究Cr3+含量对气相氟化HCFC-133a合成HFC-134a的影响。研究发现,随着活性组分Cr3+含量的增加,催化剂的活性和选择性均有所提高,如表2,当Cr3+浸渍质量分数为8.48%时,催化剂的催化性能最好,HCFC-133a的转化率达到22.6%[19]。

表2 不同Cr3+含量的催化剂催化氟化HCFC-133a合成HFC-134a的反应结果Tab 2 Reaction results of HFC-134a catalytic fluorination by HCFC-133a use catalyst with different content Cr3+
胥会祥等发现在气相氟化CH2Cl2合成CH2F2反应中,Cr含量对其催化活性具有一定的影响。Cr含量的增加,使得催化剂的孔容和比表面积增大,从而提高了反应活性,且当Cr的质量分数为10%时,其活性达到最大;但当Cr的质量分数继续增大至15%时,发现催化剂的孔容和比表面积反而减小,进而导致活性下降。这可能是因为当Cr的质量分数为10%时,催化剂表面的活性物种已经达到了单层饱和分散。因此当Cr的质量分数超过10%时,多层分散的Cr形成多聚体,反而会抑制活性物种的作用[23]。
Cho等将不同Cr含量的Cr/MgO催化剂分别用于气相氟化HCFC-133a合成HFC-134a的研究。结果发现,当Cr的质量分数从5%增加到15%时,HCFC-133a的转化率也逐渐提高,但当Cr的质量分数增加到20%时,转化率下降。Raman结果表明,催化剂中活性组分Cr含量较小 (质量分数<15%)时,形成的主要是高价态高分散的Cr物种,因此其催化活性随Cr含量的升高而提高;但当Cr含量较高(质量分数>15%)时,形成的是铬酸盐的多聚体,不利于氧原子的迁移,造成活性下降[28]。
He等研究了Cr含量对CrOx-Y2O3催化剂气相氟化合成HFC-134a反应性能的影响。研究发现,随着Cr含量的增加,HCFC-133a的转化率先增大后减小。而当Cr的质量分数为19.5%时,CrOx-Y2O3催化剂的催化活性最高,在320℃反应温度下,该催化剂对HCFC-133a的转化率达到19%。其原因是,当Cr的质量分数小于19.5%时,铬物种主要以无定形的Cr(Ⅵ)形式存在,易被氟化成活性物种CrOxFy或者Cr(OH)xFy;而当Cr含量进一步增加时,生成的是聚合态的CrOx物种,因此活性反而下降[29]。
2.5 焙烧气氛的影响
邢丽琼等用沉积-沉淀法制备了不同气氛焙烧的CrOx-Y2O3催化剂,考察了焙烧气氛对HCFC-133a气相氟化合成HFC-134a催化性能的影响。研究表明焙烧气氛对催化活性具有显著的影响,直接在空气气氛中350℃焙烧的催化剂的活性明显高于先经氮气气氛350℃焙烧和直接氮气气氛350℃焙烧的催化剂。这原因主要是直接在空气气氛下焙烧的催化剂中的Cr物种大多以CrO3的形式存在,而高价态的Cr物种有利于氟氯交换反应[30]。
Quan等分别制备了空气和氮气气氛400℃焙烧的催化剂并将其用于气相催化氟化CH2Cl2的反应。结果发现,空气气氛焙烧的催化剂的催化活性明显高于氮气气氛焙烧的催化剂。XRD和XPS结果表明空气气氛焙烧的催化剂中存在高价态的Cr物种,这可能是造成空气气氛焙烧的催化剂活性较高的原因[31]。
2.6 焙烧温度的影响
He等研究了焙烧温度对CrOx-Y2O3催化剂气相氟化HCFC-133a合成HFC-134a催化性能的影响。当反应温度为320℃时,400℃焙烧制得的催化剂对HCFC-133a的转化率为19%,而800℃焙烧得到的催化剂对HCFC-133a的转化率只有5%。此结果表明,焙烧温度明显影响了催化剂的催化性能,低温焙烧的催化剂活性高于高温焙烧的催化剂的原因主要是由于随着焙烧温度的升高,催化剂表面高分散高价态的CrO3会逐步转化为低价态聚合的Cr(Ⅴ)和Cr(Ⅲ)物种,但易被HF氟化为活性物种CrFxFy的却是高价态高分散的CrOx物种。因此焙烧温度越高,催化剂中高分散高价态的可被氟化为活性物种的CrOx物种越少,最终催化剂因活性物种的减少导致其催化性能下降[32]。
胥会祥等以CH2F2(HFC-32)的合成为目标反应,考察了焙烧温度对Cr3+/MgF2催化剂性能的影响。结果发现,随着焙烧温度的升高,催化剂的活性下降,这可能是由于焙烧温度的升高使得MgF2的晶粒尺寸增大,从而减小了催化剂的孔容和比表面积所引起的(如表 3 所示)[23]。
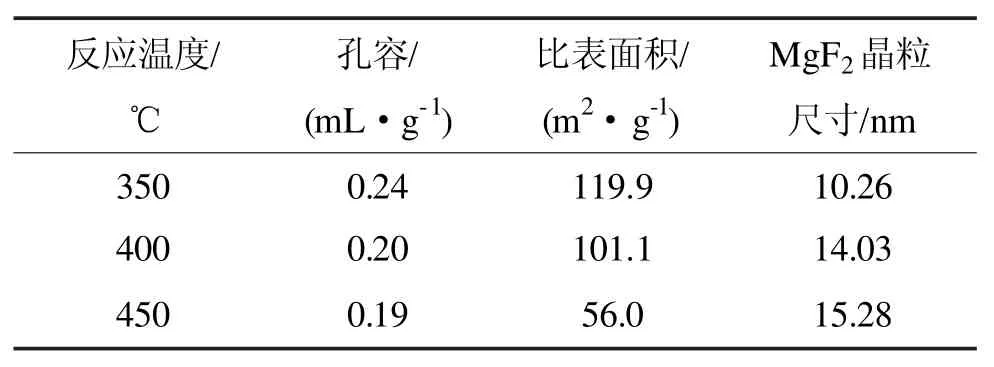
表3 不同焙烧温度下Cr3+/MgF2催化剂的孔容和比表面积Tab 3 Pore volume and specific surface area of Cr3+/MgF2 catalyst at different calcination temperature
Xie等采用沉淀法分别制备了不同温度焙烧的Cr2O3催化剂,通过气相氟化2-氯-3,3,3-三氟丙烯(HCFC-1233xy) 合成 2,3,3,3-四氟丙烯 (HFC-1234yf)反应进行研究。结果表明,焙烧温度的升高,增大了Cr2O3的晶粒尺寸,降低了催化剂的表面酸量,从而提高了催化剂的反应比速率。其中500℃下焙烧的催化剂的催化活性最高,当反应温度为320℃时,HCFC-1233xy的转化率为63.3%,HFC-1234yf的选择性为59%[33]。
Jia等考察了焙烧温度对CrOx-Y2O3催化剂气相氟化合成CF4性能的影响。研究表明,焙烧温度对催化剂的活性具有显著的影响。空气气氛下600℃焙烧的CrOx-Y2O3催化剂的活性明显高于800℃高温焙烧的催化剂[34]。一方面是因为高温焙烧的催化剂的比表面积较小,另一方面是由于低温焙烧未经氟化的催化剂中含有高价态Cr物种,而高温焙烧的催化剂中Cr物种却主要以低价态的形式存在,由于在活化过程中,只有高价态Cr物种才能转化为活性物种 CrOxFy、Cr(OH)xFy,而低价态 Cr很难被氟化,因此空气气氛下600℃焙烧的CrOx-Y2O3催化剂的活性更高。
2.7 助剂的影响
Cheng等将制备的一系列M-Cr2O3(M=Y、Co、La、Zn)催化剂用于气相氟化四氯乙烯(PCE)合成2,2-二氯-1,1,1-三氟乙烷 (HCFC-123)、2-氯-1,1,1,2-四氟乙烷(HCFC-124)和五氟乙烷(HCFC-125)的研究。研究表明,在300℃反应温度下,氟化处理后的La-Cr2O3(F)催化剂的活性最高(实验结果如表4所示),PCE的转化率达到90.6%,而HCFC-123、HCFC-124和 HCFC-125的总选择性达到93.7%[35]。这主要是因为烯烃分子较易吸附到催化剂的酸性位上,而过高的表面酸量会使催化剂的表面快速积炭,造成催化剂失活,从而使得反应的比速率降低,但在Cr2O3催化剂中添加La和Y后,催化剂表面的酸密度会降低,因此催化剂上反应的比速率增加。
胥会祥等考察了助剂(Co2+、Ni2+、Zn2+、Fe3+、Cu2+)对CrF3/MgF3催化剂催化氟化CH2Cl2性能的影响。结果发现,Co2+能提高催化剂的稳定性,Ni2+可减少积炭,从而延长催化剂的寿命,Zn2+能提高CH2F2的选择性,而Fe3+和Cu2+却会使催化剂的活性下降[36]。
毛汉卿等研究了Cr-Mg-Zn催化剂在气相氟化合成HFC-134a反应中的催化性能。结果发现,Zn的加入导致催化剂形成少量结晶,使催化剂强度增强,从而提高了催化剂的寿命和稳定性[37];董洪涛等将Cr-Zn-Al催化剂同样用于气相氟化合成HFC-134a反应中,发现在Cr中添加Zn后,虽然降低了催化剂的比表面积,但提高了催化剂的强度,使得催化剂的稳定性和寿命均有所提高,同时,添加的Al活化后可能形成了CrAlOxFy物种,可以有效地断裂C—Cl键,生成 C—F键,进而提高催化剂的催化性能[38]。
Bonniface等通过同位素标记法研究了添加Zn或Ni后的Cr2O3催化剂对气相氟化HCFC-133a合成HFC-134a反应性能的影响。结果表明,在Cr2O3催化剂中添加少量Zn或Ni后可以提高催化剂的活性,但随着添加量的进一步增加,反而会导致活性下降。产生这一现象的原因主要是因为添加的Zn或Fe会直接与催化剂的活性位相连,一方面会降低HF吸附在表面的活化能,另一方面,添加的Zn或Fe还可以提供其他的活性位,或提供一种能移走催化剂表面吸附的Cl原子的途径,这几方面的作用使得催化剂的活性有所提高,然而当进一步增加Zn或Ni的含量时,催化剂的活性中心会减少,因此过多助剂的添加反而导致催化剂的活性中心减少,活性降低[39]。
Zhu等发现V、Zr助剂的掺杂使得铬基催化剂的比表面积和催化活性下降,而Mg助剂的加入则会增大铬基催化剂的比表面积,从而提高其活性和选择性[40]。
3 铬基催化剂的失活原因
3.1 活性组分流失
对于铬基催化剂而言,由于催化剂中高价态铬物种的沸点较低,因此当反应温度较高时,活性组分容易流失,从而造成催化剂活性下降。
邢丽琼等研究发现,直接在空气气氛下焙烧得到的催化剂在气相氟化HCFC-133a合成HFC-134a反应中,初始转化率可达到35%,但10 h后活性便开始降低,65 h后转化率仅为15%。这主要是催化剂中高价态Cr物种易流失,从而导致催化剂的稳定性下降[30]。
Albonetti等同样认为,CrO3在氟化过程中生成的CrO2F2容易挥发,从而导致Cr物种流失,造成活性降低。大量研究表明,高价态铬物种易转变为活性物种,因此有利于提高反应的活性,但却容易在反应过程中流失,导致催化剂的稳定性下降;而低价态铬物种虽不易流失,但催化活性低,因此如何获得既有高活性,又具有高稳定性的催化剂是目前氟化工行业中较为关注的问题[41]。
3.2 晶相转变
催化剂的晶相转变主要原因是过高的反应温度所造成的。当反应温度较高时,催化剂的载体和活性组分都有可能发生晶相转变,从而造成失活现象。
吕剑等将CrF3/AlF3催化剂用于气相氟化合成HFC-134a的强化实验中进行研究。结果发现,在400℃反应温度下,CrF3/AlF3催化剂经过1 000 h的强化实验后,载体AlF3的晶粒明显增大,比表面积随之下降,而其晶型也从γ-AlF3转变成了α-AlF3,但由于α-AlF3无明显的催化活性,最终导致催化剂的活性明显下降[19]。
3.3 表面烧结
在一些强放热的反应中,如果控制不得当,易导致反应热无法及时地转移,从而造成局部迅速升温,催化剂表面烧结的现象,其中可能包括晶相转变与熔融,最终使得催化剂失活。
Albonetti等发现,CrOxFy/AlF3催化剂在气相氟化合成HFC-134a反应中会发生烧结现象。XRD结果发现,当反应温度从650℃上升到800℃时,载体从 γ-AlF3相继转变为 β-AlF3、δ-AlF3, 最后变为 α-AlF3,然而α-AlF3没有明显的催化活性,最终造成催化剂失活[41]。
3.4 催化剂积炭
由于气相氟氯交换反应的反应温度较高,所以容易导致反应原料和产物间发生脱卤化氢作用,产生烯烃类副产物,而烯烃类产物会进一步聚合形成积炭,造成催化剂的表面被污染,孔道被堵塞,从而降低催化活性。
Lee等发现在气相氟化CF3CH2Cl反应中,当反应温度分别为400、450℃时,随着反应时间的增加,Cr-Mg氟化催化剂逐渐失活。失活原因主要是CF3CH2Cl在高温下易发生消去反应,生成CF2=CHCl后再发生聚合,最终产生积炭导致催化剂失活[42]。
钱林等在气相氟化CCl2F2合成CF4的反应中发现,在反应温度为400℃时,CrOx-Y2O3催化剂对CCl2F2的转化率在97%左右,且反应基本处于稳定状态[5];He等将 CrOx-Y2O3催化剂用于气相氟化CF3CH2Cl(HCFC-133a)合成 CF3CH2F(HFC-134a)反应,研究发现,在320℃反应温度下,HCFC-133a的转化率一直稳定在19%左右[32];而Xie等发现,在气相氟化CH2=CClCF3(HCFC-1233xf)合成 CH2=CFCF3(HFC-1234yf)反应中,以 500 ℃焙烧的 Cr2O3作为催化剂,结果发现,在320℃下,随着反应的进行,HCFC-1233xy的转化率逐渐下降 (从65%下降至30%),这可能是由于HCFC-1233xy含碳数多,易产生积炭从而造成催化剂失活,进而降低了反应的活性和稳定性[33]。
孙万付等用实验证明了不同烷烃、不同结构的烃以及不同烯烃对催化剂积炭有一定的影响[43]。烷烃的碳链越长,积炭量就越多,这主要是因为裂化反应依据的是碳正离子反应机理,因此当直链烷烃的结构一致而含碳数目不同时,碳链较长的烷烃和催化活性中心接触的概率更大,导致反应生成的碳正离子也更多,最终使得裂化反应速率更快。随后,生成的碳正离子又继续发生反应,比如发生热消除反应,生成小分子烯烃,而产生的烯烃又和碳正离子或是其他化合物发生异构化反应和缩合反应,产生焦炭前身体,然后焦炭前身体再进一步发生脱氢反应或缩合反应,最后形成焦炭,因此催化剂的积炭量随碳链的增长而增多。
另外,烃的结构不同,积炭难易程度也有差别,例如,正戊烷没有支链和不饱和键,且空间结构简单,因此积炭量很少;而异戊烯和异戊二烯的双键都在端基位,和催化剂的活性中心较易发生作用,所以相比正戊烷更易积炭。又因为异戊二烯的双键多,反应概率大,故其积炭量又比异戊烯多。这也表明催化剂的积炭很可能来自烯烃的聚合反应。
然而,对于不同的烯烃,碳链越长,积炭量却越少,比如,辛烯与十二烯都是α烯烃,双键都在端基位,由于催化剂的活性中心会先吸附不饱和位,故当碳链较长时,空间阻碍效应更大,从而造成双键位和其他化合物发生聚合反应形成积炭的概率降低,其积炭量减少。
一般情况下,积炭是导致催化剂失活的主要原因,通常采用氧化烧炭法使催化剂再生,具体方法为在300~500℃高温下,将氧气通入催化剂床层,通过有效燃烧以除去催化剂表面的积炭,但由于燃烧过程中会放出大量的热造成催化剂烧结,所以必须控制好氧分压,起初的氧含量较低,随后逐渐增加,为了控制好氧含量,通常用氮气进行稀释。
4 展望
气相氟氯交换反应是合成氟代烃较为理想的路线,但也仍然面临一些挑战。催化剂失活是面临的一大难题。为了延缓积炭,通常在使用过程中,在原料气中加入氮气进行稀释,再采用高温氧气再生法除去催化剂表面的积炭,但由于再生过程中释放的热量会导致催化剂表面烧结,因此要严格控制再生气中的氧含量。
而铬基催化剂作为气相氟氯交换反应的主体催化剂,虽然已有大量的研究和应用,但也存在挑战,比如铬基催化剂中高价态铬物种的活性高但却易流失,导致催化剂的稳定性下降,而低价态铬虽不易流失,但活性较低,因此如何获得高活性、高稳定性的铬基催化剂成为氟化工行业中非常关注的问题。
目前,对铬物种在氟化过程中的转化,反应的活性中心以及铬基催化剂表面物种与氟氯交换反应性能间关系的研究也显得尤为重要。因此,在真正认识反应的活性中心和反应机制的基础上,提高铬基催化剂的抗积炭、抗烧结能力,加强催化剂的再生工艺,提高催化剂的催化活性,以及延长催化剂的使用寿命等将成为今后氟化工领域研究的方向。
[1]李惠黎,任建刚.环保型制冷剂——氢氟烃的生产、性质及应用[M].北京:化学工业出版社,2003:1-31.
[2]Fisher D A,Midgley P M.The production and release to the atmosphereofCFCs113,114,115[J].AtmosphericEnvironment.Part A:General Topics,1993,27(2):271-276.
[3]Didion D A,Bivens D B.Role of refrigerant mixtures as alternatives to CFCs[J].International Journal of Refrigeration,1990,13(3):163-175.
[4]Busenberg E,Plummer L N.Use of chlorofluorocarbons(CCl3F and CCl2F2)as hydrologic tracers and age-dating tools:The alluvium and terrace system of central Oklahoma[J].Water Resources Research,1992,28(9):2257-2283.
[5]钱林.气相氟化合成CFCs替代品的Cr基催化剂的表征与性能研究[D].金华:浙江师范大学,2005.
[6]Cheminal B,Lacroix E,Lantz A,et al.Process for fluorination of perchloroethylene or of pertachloroethane:US,5932776[P].1999-08-03.
[7]Yoshimura T,Homoto Y,Yamada Y,et al.Process for producing pentafluoroethane and tetrafluoroethane:US,6011185[P].2000-01-04.
[8]Malay M D.Process for preparing 2,2,3,3-tetrafluoropropene:US,2931840[P].1960-04-05.
[9]Rao V N,Mallikarjuna,Sievertown Allen C.Process for preparing 2,3,3,3-tetrafluoropropene:WO,2008060614A2[P].2008-05-22.
[10]Unveren E,Kemnitz E,Lippitz A,et al.Suface characterization of chromia for Chlorine/Fluorine exchange reactions[J].Journal of Physical Chemistry B,2005,109:1903-1913.
[11]Steven C Y,David F C.Dehalogenation of 1,1,2-trichloro-1-fluoroethane over-Cr2O3[J].Journal of Physical Chemistry B,2003,107:5182-5189.
[12]吕剑,石磊,李惠黎,等.合成 CFC-12替代物 HFC-134a的CrF3/AlF3催化剂研究[J].高等学校化学学报,1998,19(10):1677-1679.
[13]Rao J M,Sivaprasad A,Rao P S,et al.A comparative study of bulk and supported chromia catalysts for the fluorination of trichloroethylene[J].Journal of Catalysis,1999,184:105-111.
[14]Alonso C,Morato A,Medina F,et al.Effect of the aluminium fluoride phase for the Cl/F exchange reactions in CCl2F2(CFC-12)and CHClF2(HCFC-22)[J].Applied Catalysis B:Environmental,2003,40:259-269.
[15]Cho D H,Kim Y G,Chung M J,et al.Preparation of characterization of magnesia-supported chromium catalysts for the fluorination of 1,1,1-trifluoro-2-chloroethane(HCFC-133a)[J].Applied Catalysis B:Environmental,1998,18:251-261.
[16]Lee H,Kim H S,Kim H,et al.Preparation of 1,1,1,2-tetrafluoroethane by catalytic fluorination of 1,1,1-trifluo ro-2-chloroethane over CrF3/MgF2-AlF3.Journal of Molecular Catalysis A:Chemical,1998,136:85-89.
[17]Sekiya A,Quan H D,Tamura M,et al.Fluorination of chorofluoroetherusing catalystssupported by porous aluminum fluoride[J].Journal of Fluorine Chemistry,2001,112:145-148.
[18]Wang Fang,Fan Jinglian,Zhao Yang,et al.Effects of yttrium-doping on the performance of Cr2O3catalysts for vapor phase fluorination of 1,1,2,3-tetrachloropropene[J].Journal of Fluorine Chemistry,2014,166:78-83.
[19]吕剑,石磊,杨会娥,等.合成 CFC-12替代物 HFC-134a用CrF3/AlF3催化剂的研究[J].催化学报,1997,18(5):388-391.
[20]张学良.气相氟化法合成二氯甲烷的研究进展[J].化工生产与技术,2004,11(6):7-10.
[21]牛怀成,李利春,李瑛,等.高比表面积氟化镁的合成及其在催化中的应用研究进展[J].化工进展,2012,31(7):1484-1492.
[22]罗孟飞,王芳,范镜莲,等.用于HFC-245fa裂解联产HFC-1234ze和HFC-1234yf的催化剂及其制备方法:中国,103537305A[P].2014-01-29.
[23]胥会祥,吕剑.Cr3+/MgF2氟化催化剂的制备及其对合成二氟甲烷反应的催化性能[J].催化学报,2003,24(5):379-384.
[24]Cho D H,Kim Y G,Chung M J,et al.Preparation and characterization of magnesia-supported chromium catalysts for the fluorination of 1,1,1-trifluoro-2-chloroethane(HCFC-133a)[J].Applied Catalysis B:Environmental,1998,18:251-261.
[25]Rao J M,Sivaprasad A,Rao P S,et al.A comparative study of bulk and supported chromia catalysts for the fluorination of trichloroethylene[J].Journal of Catalysis,1999,184:105-111.
[26]钱林,邢丽琼,毕庆员,等.气相氟化合成1,1,1,2-四氟乙烷的CrOx-Y2O3催化剂的表征与性能[J].物理化学学报,2009,25(2):336-340.
[27]Jia Wenzhi,Jin Lingyun,Wang Yuejuan,et al.Fluorination of dichlorodifluoromethane to synthesize tetrafluoromethane over Cr2O3-AlF3catalyst[J].Journal of Industrial and Engineering Chemistry,2011,17:615-620.
[28]Cho D H,Kim Y G,Chung J S.Catalytic fluorination of 1,1,1-trifluoro-2-chloroethane(HCFC-133a)over chromium catalysts[J].Catalysis Letters,1998,53:199-203.
[29]He Jun,Lu Jiqing,Xie Guanqun,et al.Characterization of CrOx-Y2O3catalysts for fluorination of 2-chloro-1,1,1-trifluoroethane[J].Indian Journal of Chemistry,2009,48A:489-497.
[30]邢丽琼,钱林,毕庆元,等.CrOx-Y2O3催化剂中Cr物种对氟氯交换反应性能的影响[J].物理化学学报,2009,25(9):1928-1932.
[31]Quan H D,Tamura M,Matsukawa Y,et al.Investigation into chromia-based catalyst and its application in preparing difluoromethane[J].JournalofMolecularCatalysisA:Chemical,2004,219:79-85.
[32]He Jun,Xie Guanqun,Lu Jiqing,et al.Effect of calcination temperature on CrOx-Y2O3catalysts for fluorination of 2-chloro-1,1,1-trifluoroethane to 1,1,1,2-tetrafluoroethane[J].Journal of Catalysis,2008,253:1-10.
[33]Xie Zunyun,Fan Jinglian,Cheng Yongxiang,et al.Cr2O3catalysts for fluorination of 2-chloro-3,3,3-trifluoropropene to 2,3,3,3-tetrafluoropropene[J].Industrial and Engineering Chemistry Research,2013,52:3295-3299.
[34]Jia Wenzhi,Xing Liqiong,Qian Lin,et al.CrOx-Y2O3catalysts for vapor phase fluorination of dichlorodifluoromethane[J].Indian Journal of Chemistry,2009,49A:1212-1216.
[35]Cheng Yongxiang,Fan Jinglian,Xie Zunyun,et al.Effects of M-promoter(M=Y,Co,La,Zn)on Cr2O3catalysts for fluorination of perchloroethylene[J].Journal of Fluorine Chemistry,2013,156:66-72.
[36]胥会祥,吕剑.MgF2基催化剂合成CH2F2的研究[J].工业催化,2002,10(5):50-53.
[37]毛汉卿,祝俊丽,赵翀.Cr-Mg-Zn催化剂气相氟化合成1,1,1,2-四氟乙烷的研究[J].浙江化工,2007,38(4):3-5.
[38]董洪涛,陈纪忠.Cr-Zn-Al催化剂气相氟化法合成HFC-134a[J].精细化工,2006,23(2):138-140.
[39]Bonniface D W,Scott J D,Watson M J,et al.Halogen exchange reactions for CFC alternatives:the behaviour of fluorine-18 labeled hydrogen fluoride towards prefluorianted chromia containing nickl(Ⅱ)or zinc(Ⅱ)[J].Green Chemistry,1999,1:9-11.
[40]Zhu Y,Fiedler K,Rüdiger St,et al.Aliovalent-substituted chromium-based catalysts for the hydrofluorination of tetrachloroethylene[J].Journal of Catalysis,2003,219:8-16.
[41]Albonetti S,Forni L,Cuzzato P,et al.Aging investigation on catalysts for hydrofluorocarbons synthesis[J].Applied Catalysis A:General,2007,326:48-54.
[42]Lee H,Jeong H D,Chung Y S,et al.Fluorination of CF3CH2Cl over Cr-Mg fluoride catalyst:the effect of temperature on the catalyst deactivation[J].Journal of Catalysis,1997,69:307-316.
[43]孙万付,马波,索继栓,等.加氢裂化催化剂积炭行为的研究[J].催化学报,2000,21(3):269-272.
猜你喜欢
化工进展(2023年1期)2023-03-01
云南化工(2020年11期)2021-01-14
汽车维护与修理(2018年7期)2018-10-13
浙江大学学报(工学版)(2016年11期)2016-06-05
中国卫生标准管理(2015年17期)2016-01-20
合成技术及应用(2015年2期)2016-01-10
化学反应工程与工艺(2015年1期)2015-04-16
中国当代医药(2015年9期)2015-03-01
装备环境工程(2015年4期)2015-02-28
中国塑料(2014年12期)2014-10-17
